

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Acute myeloid leukemia (AML) is the second most common disease among all hematologic malignancies and the most common disease in adult myeloid neoplasms. Advanced genomics studies conducted using hematopoietic stem cells or progenitors have found that the disease is characterized by pathogenic heterogeneity and simultaneous alterations in multiple genes.
For decades, the treatment paradigm for AML—the achievement of complete remission (CR) with anthracycline and cytarabine combined induction chemotherapy followed by allogeneic hematopoietic stem cell transplantation (HSCT)—has remained unchanged. Treatment strategies could be modified by risk groups according to cytogenetic profiles including promyelocytic leukemia/retinoic acid receptor alpha (PML/RARA), core-binding factor subunit beta/myosin heavy chain 11 (CBFb/MYH11), runt-related transcription factor1/runt-related transcription factor1 translocation partner 1 (RUNX1/RUNX1T1) translocation that were known before the next-generation sequencing (NGS) era, however, only to decide whether to give several cycles of consolidation chemotherapy or to undergo HSCT.
Due to the toxicity of the high-intensity treatment, it could only be applied to young and fit patients. AML is a well-known age-related disease. About half of AML patients are diagnosed at an age older than 70 years. Even though hypomethylating agents were developed around 20 years ago for the treatment of elderly AML patients, there has been little improvement in clinical outcomes and survival rates.1
Since 2010, with the remarkable development of genomics and sequencing technology, even normal karyotypes of AML, accounting for over half of the total, mutations were frequently discovered in genes, including fms related receptor tyrosine kinase 3 (FLT3), nucleophosmin 1 (NPM1), RAS, deoxyribonucleic acid methyltransferase 3A (DNMT3A), CCAAT enhancer binding protein alpha (CEBPA), isocitrate dehydrogenase 1/2 (IDH1/2), and tet methylcytosine dioxygenase 2 (TET2).2 Based on this genomic information, novel therapeutic targets and drugs have been developed, including an FLT3 inhibitor that acts on a tyrosine kinase receptor and an IDH1/2 inhibitor that interrupts epigenetic changes. Recently, along with the advancement of cancer immunotherapy, chimeric antigen receptor T cell (CAR-T) therapy targeting AML cells is undergoing active development.3 The treatment strategy for AML is changing from being limited to only 2 options (cytotoxic chemotherapy followed by HSCT or hypomethylating agent) to the availability of various novel target therapies.
Genetic mutations in AML can be classified into transcription factors, kinases, cell cycle regulators, spliceosomal genes, and epigenetic regulators.4 Of these, mutations in epigenetic regulators are the most frequent, implying that epigenetic dysregulation is critical in AML pathogenesis.4 Epigenetic changes are inherited DNA alterations that do not change the DNA sequence. Epigenetic alterations are classified into DNA methylation, histone modification, and chromatin remodeling. It is well known that altered gene expression caused by epigenetic dysregulations is an important carcinogenic mechanism.56 Furthermore, some epigenetic changes are reversible and can become novel therapeutic targets.7 A recent NGS study reported that more than 70% of AML patients showed mutations at genes related in DNA methylation or histone acetylation.27
For this reason, most new drugs for AML are mainly focused on the epigenetic modifications, except FLT3 inhibitors and CAR-T therapy. Summary of targetable alterations and novel agents are described in Fig. 1. Generally, the therapeutic efficacies of epigenetic agents as single agents are limited.8 We review focused on the clinical perspective of epigenetic agents and combination therapy among emerging novel therapies for AML with the advanced NGS technology.
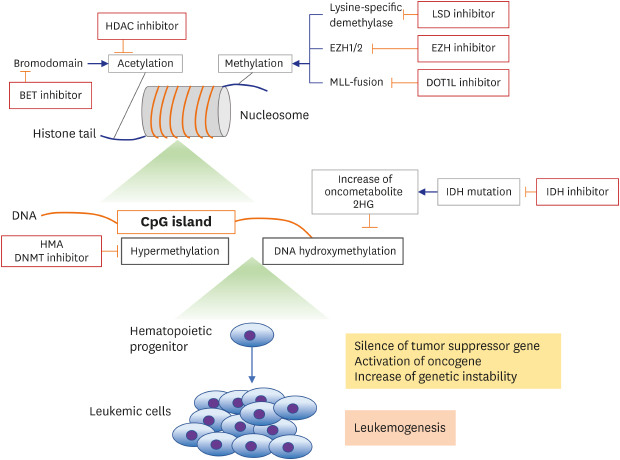
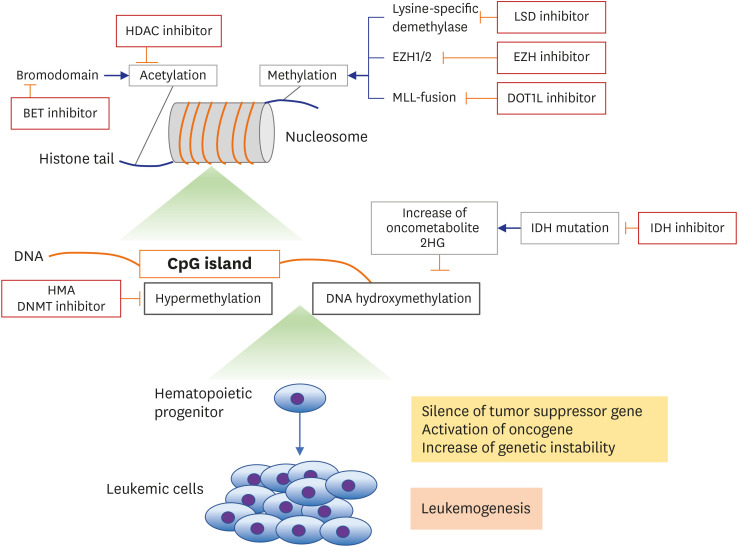
Fig. 1
Overview of epigenetic mechanisms and targeting agents.
HDAC = histone deacetylase, EZH = enhancer of zeste 2 polycomb repressive complex, LSD = lysine demethylase, MLL = myeloid/lymphoid or mixed-lineage leukemia, DOT1L = DOT1 like histone lysine methyltransferase, HMA = hypomethylating agent, DNMT = DNA methyltransferase, IDH = isocitrate dehydrogenase.
MONOTHERAPIES
DNA methyltransferase (DNMT) inhibitors
Many researchers have investigated the correlation of DNA methylation with carcinogenesis and progression.9 DNMT inhibition is a major treatment strategy for AML patients who are not feasible for intensive chemotherapy. DNMT adds a methyl group to the carbon-5 position of cytosine ring from S-adenosyl methionine (SAM) to form S-adenosyl-l-homocysteine (SAH), thus resulting in the regulation of gene expression.6910 In carcinogenesis, DNMT aberration hypermethylates tumor suppressor genes (TSG) and repress their expression. Hence, inhibition of DNMT through hypomethylation can restore normal molecular function.41112
The most investigated DNMT inhibitors are hypomethylating agents (HMAs), including azacitidine and decitabine. Currently, 2 azanucleosides that are pyrimidine analogues of cytidine, azacitidine, and decitabine are used as treatments for AML and myelodysplastic syndrome (MDS) patients who are not eligible for intensive chemotherapy.1113 These agents incorporate into DNA (decitabine) or RNA (azacitidine) and cause degradation of DNMT and DNA hypomethylation.713 Decitabine and azacitidine can also cause direct DNA damage and promote the expression of tumor-associated antigens, thus eliciting an anti-leukemic immune reaction.1113 Furthermore, azacitidine incorporates into RNA and interrupts protein synthesis and induces apoptosis.13
Azacitidine 75 mg/m2 for 7 days repeated every 28 days has been approved since 2004,1415 and decitabine 20 mg/m2 for 5 days repeated every 28 days has been approved since 2006 by United States Food and Drug Administration (FDA).1617 In a phase III international randomized study, AZA-001, the hematological improvement in AML and MDS patients was 47% and the overall survival (OS) in azacitidine group was 24.5 months, while that in the best supportive care (BSC) group was 15 months.15 As the efficacy of azacitidine in MDS was established, its efficacy in AML was studied because patients with bone marrow (BM) blast proportion up to 30% were included in the AZA-001 trial and the subgroup analysis showed a significant survival benefit.18 Unfortunately, patients with blast proportions > 30% did not meet the primary endpoint of a clinical trial comparing azacitidine and conventional care, even though they showed a tendency of improved OS.19
Decitabine is an azacitidine analogue that is incorporated into newly synthesized DNA strands.7 In phase III trials, decitabine showed prolonged progression-free survival, but the difference of OS was not significantly different.17 The efficacy of decitabine was also studied in AML patients, compared to BSC or low-dose cytarabine. Decitabine showed higher response rates (overall response rate [ORR], 64%; CR, 50%) and improvement of quality of life, but nonsignificant increase in median OS.20 Both agents are known to improve hematological response and quality of life. The difference between the 2 agents is based on the result of a retrospective study that compared azacitidine and decitabine in older patients and reported that azacitidine had a longer OS.21 With developing novel strategies, intensified HMA regimens of 10-day (decitabine) or 5-day in every 14 days (azacitidine) showed improved response rates in high-risk patients.13 Due to toxicities, their clinical trials must be meticulously designed.
New HMAs, including oral form of azacitidine and decitabine, are under investigation with promising response rate and tolerable toxicities. Guadecitabine,22 a second-generation DNA methylation inhibitor, is a dinucleotide of decitabine and deoxyguanosine. While the CR rate of HMAs is no better than 30%, guadectiabine showed 57% CR with 18.2 months of OS in a newly diagnosed elderly AML patient. In a recent phase II trial, guadecitabine presented 51% and 43% of response rate in each treatment-naïve patients and refractory patients.2324 However, recent ongoing phase III trials that evaluating guadecitabine in unfit treatment naïve and relapse/refractory AML patients were announced failed to meet primary endpoints of CR and prolonged survival rate.13 A phase II study of guadecitabine to evaluate the CR rate in high risk MDS is ongoing.
These nucleoside analogues reverse DNA hypermethylation and do not have DNMT-specific activities. Novel DNMT3A-specific target molecules are currently under development.69 Suggested mechanisms for DNMT inhibition include targeting the catalytic domain, interruption of DNA and enzyme binding, DNA binding, DNA and SAM competition, structural allosteric inhibition, and inhibition of protein-protein interactions.25 Most of these agents are still experimental. Thus, novel potent target agents need to be developed urgently.
IDH1/2 inhibitors
Isocitrate dehydrogenases (IDHs) have 2 isotypes (IDH1 and IDH2) and are present in 15%–30% of AML patients.26 The normal functions of IDH include adaptation to hypoxia, histone demethylation, and DNA modification, by catalyzing the conversion of isocitrate to α-ketoglutarate.827 If mutations occur in IDH1 and IDH2, α-ketoglutarate is converted to 2-hydroxyglutarate (2-HG) in a nicotinamide adenine dinucleotide phosphate hydrogen-dependent manner. Increased 2-HG leads to gene hypermethylation, resulting in enhanced cellular proliferation, aberrant gene expression, and the inhibition of myeloid differentiation.22 Frequent co-occurrence of IDH and DNMT3A mutations is reported.26 More interestingly, 2-HG competitively inhibits tet methylcytosine dioxygenase 2 (TET2) protein which regulates DNA methylation. TET2 loss-of-function mutation is frequently found in AML. Therefore, co-occurrence of IDH and TET2 mutations leads to global DNA hypermethylation, thus contributing to leukemogenesis.262728
At present, 2 IDH inhibitors have been approved by the US FDA: the IDH1 inhibitor ivosidenib (AG-120) and the IDH2 inhibitor enasidenib (AG-221). Both are oral small molecules that inactivate mutated IDH proteins via allosteric inhibition and reduction of the oncometabolite 2-HG.2729
Ivosidenib showed tolerable toxicity at 500 mg once daily, with a low frequency of grade 3 or higher adverse events in the phase 1 trial.30 Remission rates were 41.6%, with a 6.5-month treatment duration in relapsed-refractory AML.31 Enasidenib showed an ORR of 38.8% with a 8.8-month treatment duration at 100 mg once daily in relapsed-refractory AML. A potentially fatal adverse effect of both IDH inhibitors is differentiation syndrome, occurs in approximately 40% of patients. Similarly, with differentiation syndrome in acute promyelocytic leukemia treated with all-trans retinoic acid, pulmonary infiltrates, pleural effusion, fever and hypotension and respiratory distress can be observed. The median time of onset is 20 days for ivosidenib and 19 days for enasidenib. The relative risk is higher when the BM blast proportion exceeds 48%. Clinical outcomes, including duration of remission and OS, were inferior in patients who experienced differentiation syndrome. However, since patients with differentiation syndrome were more likely to have less than 80% of ivosidenib or enasidenib dose intensity, a correlation cannot be drawn between differentiation syndrome and poor prognosis.32
Histone deacetylase (HDAC) inhibitors
Histone acetylation is a highly dynamic process regulated by histone acetyltransferases and HDACs, via the modification of gene transcription and expression.8 DNA wraps around histones, consisting of 4 core types (2A, 2B, 3, and 4) and wrapped in a unit called nucleosome.33 The N-terminal region of histone tails plays major roles: histone modification during transcription, chromatic structure, and DNA repair.263435 Acetylation of the lysine residues in the N-terminal region leads to gene expression by enabling DNA binding to transcription factors.33 On the other hand, histone deacetylation is the reverse of lysine acetylation via HDACs and gene transcription reduction.34
Histone acetylation levels are well known to be altered in cancer. Chimeric chromosomal fusion abnormalities in leukemia, including PML-RARA, promyelocytic leukemia zinc finger-RARA (PLZF-RARA), and AML1-ETO, increase HDAC levels and suppress gene expression, thus contributing to leukemogenesis.36 Deacetylation of P53, a well-known TSG, induces the suppression of P53-dependent growth arrest and apoptosis, and increases leukemogenesis risk. Interestingly, mutations of HDAC genes have not been found in AML.33 However, other genes that affect HDACs, including enhancer of zeste 2 polycomb repressive complex 2 subunit (EZH2) and additional sex combs like 1, transcriptional regulator 1 (ASXL1), are commonly mutated in AML.3437
HDAC inhibitors increase histone acetylation to reactivated TSGs, and most are still under development for AML treatment; however, several agents have been FDA approved for other hematologic malignancies. HDAC inhibitors are categorized into 2 groups: HDAC isoform selective inhibitors and pan-HDAC inhibitors.33 Till date, 4 HDAC inhibitors have been approved by the US FDA: vorinostat for cutaneous T-cell lymphoma (CTCL),35 panobinostat for multiple myeloma,38 belinostat for peripheral T-cell lymphoma (PTCL),39 and romidepsin for CTCL and PTCL.40 Vorinostat, panobinostat, and belinostat are pan-HDAC inhibitors, while romidepsin is a selective inhibitor. They all demonstrated inhibitory effects against AML cells in vitro and in vivo. Even though promising results of preclinical researches, vorinostat, panobinostat and belinostat showed minimal activities in clinical studies targeting advanced AML.3334 The disappointing clinical activity was similar in romidepsin. The therapeutic potential of HDAC inhibitors as monotherapy is limited; thus, their application to combination therapy with other therapeutic options such as cytotoxic chemotherapy and target agents, is being investigated.
Other agents
Besides the many epigenetic agents mentioned above, many novel target molecules are in clinical development, including the DOT1 like histone lysine methyltransferase (DOT1L) inhibitors (EPZ004777, EPZ-5676), bromodomain and extra-terminal domain (BET) bromodomain inhibitor, lysine demethylase 1 (LSD1) inhibitor, EZH inhibitor, and B-cell chronic lymphocytic leukemia/lymphoma 2 (BCL-2; venetoclax) inhibitors.822 The suppression of Hox expression by DOT1L inhibitors results from the rearrangement of the myeloid/lymphoid or mixed-lineage leukemia (MLL) gene, and was responsible for its observed antileukemic activities in early preclinical and early phase clinical research; however, this efficacy was minimal in later phase trials.84142 BET inhibitors inhibit the interaction between MLL fusion proteins and BET bromodomains, suppressing apoptotic BCL2 and c-MYC genes.43 Like other epigenetic small molecules, BET inhibitors reportedly showed no promising results in human trials.44 LSD1 inhibitors, which induce leukemic cell differentiation in animal models, and EZH inhibitors, which methylate histones and repress gene transcription, both failed to exhibit adequate safety and efficacy in human trials.45 The BCL-2 inhibitor, venetoclax, is a notable novel hematologic malignancy agent, already approved for chronic lymphocytic leukemia.46 Venetoclax was approved in combination with hypomethylating agents or low-dose cytarabine, in elderly AML, but not as a monotherapy.464748 Clinical trials of epigenetic agents as monotherapies are described in Table 1.
Table 1
Selected epigenetic target agents in monotherapy clinical trials
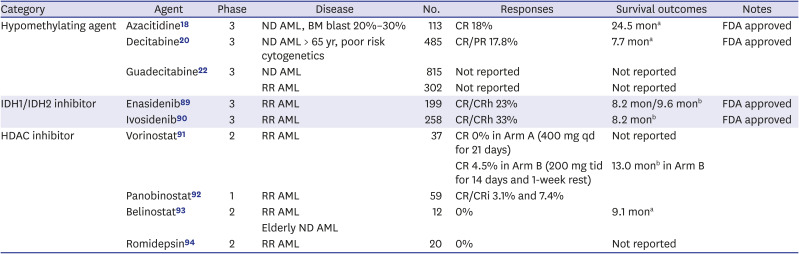
| Category | Agent | Phase | Disease | No. | Responses | Survival outcomes | Notes |
|---|---|---|---|---|---|---|---|
| Hypomethylating agent | Azacitidine18 | 3 | ND AML, BM blast 20%–30% | 113 | CR 18% | 24.5 mona | FDA approved |
| Decitabine20 | 3 | ND AML > 65 yr, poor risk cytogenetics | 485 | CR/PR 17.8% | 7.7 mona | FDA approved | |
| Guadecitabine22 | 3 | ND AML | 815 | Not reported | Not reported | ||
| RR AML | 302 | Not reported | Not reported | ||||
| IDH1/IDH2 inhibitor | Enasidenib89 | 3 | RR AML | 199 | CR/CRh 23% | 8.2 mon/9.6 monb | FDA approved |
| Ivosidenib90 | 3 | RR AML | 258 | CR/CRh 33% | 8.2 monb | FDA approved | |
| HDAC inhibitor | Vorinostat91 | 2 | RR AML | 37 | CR 0% in Arm A (400 mg qd for 21 days) | Not reported | |
| CR 4.5% in Arm B (200 mg tid for 14 days and 1-week rest) | 13.0 monb in Arm B | ||||||
| Panobinostat92 | 1 | RR AML | 59 | CR/CRi 3.1% and 7.4% | |||
| Belinostat93 | 2 | RR AML | 12 | 0% | 9.1 mona | ||
| Elderly ND AML | |||||||
| Romidepsin94 | 2 | RR AML | 20 | 0% | Not reported |
ND = newly diagnosed, AML = acute myeloid leukemia, BM = bone marrow, CR = complete remission, FDA = Food and Drug Administration, PR = partial remission, RR = relapse and refractory, CRh, CR with partial platelet recovery, HDAC = histone deacetylase, qd = once a day, tid = three times a day, CRi, CR with incomplete platelet recovery.
aMedian overall survival; bMedian duration of response.
COMBINATION THERAPY
The inefficacy of the above epigenetic agents in monotherapy implies that treatment with a single epigenetic agent is insufficient for the attaining of clinical benefits in AML. AML is one of the most heterogenous malignancies genetically, with enormous altered genetic events that affect hematopoietic progenitors or stem cells. Even in the genomics era, target agent monotherapy is insufficient for complete leukemic cell extinction, and conventional cytotoxic chemotherapy followed by HSCT still has a curative role in AML. However, most of the novel small molecules which were unsuccessful as monotherapies showed synergistic effects in combination with hypomethylating agents, conventional chemotherapy, and other target drugs. Therefore, it is deducible that the answer of new treatment strategies for next era might be the combination. Most of combination trials are undergoing based on HMAs (Table 2).
Table 2
Selected epigenetic target agents in combination therapy clinical trials
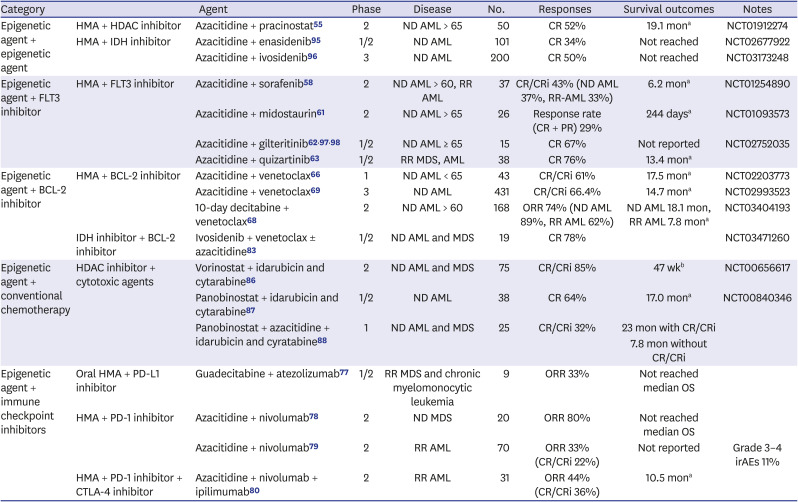
| Category | Agent | Phase | Disease | No. | Responses | Survival outcomes | Notes | |
|---|---|---|---|---|---|---|---|---|
| Epigenetic agent + epigenetic agent | HMA + HDAC inhibitor | Azacitidine + pracinostat55 | 2 | ND AML > 65 | 50 | CR 52% | 19.1 mona | NCT01912274 |
| HMA + IDH inhibitor | Azacitidine + enasidenib95 | 1/2 | ND AML | 101 | CR 34% | Not reached | NCT02677922 | |
| Azacitidine + ivosidenib96 | 3 | ND AML | 200 | CR 50% | Not reached | NCT03173248 | ||
| Epigenetic agent + FLT3 inhibitor | HMA + FLT3 inhibitor | Azacitidine + sorafenib58 | 2 | ND AML > 60, RR AML | 37 | CR/CRi 43% (ND AML 37%, RR-AML 33%) | 6.2 mona | NCT01254890 |
| Azacitidine + midostaurin61 | 2 | ND AML > 65 | 26 | Response rate (CR + PR) 29% | 244 daysa | NCT01093573 | ||
| Azacitidine + gilteritinib629798 | 1/2 | ND AML ≥ 65 | 15 | CR 67% | Not reported | NCT02752035 | ||
| Azacitidine + quizartinib63 | 1/2 | RR MDS, AML | 38 | CR 76% | 13.4 mona | |||
| Epigenetic agent + BCL-2 inhibitor | HMA + BCL-2 inhibitor | Azacitidine + venetoclax66 | 1 | ND AML < 65 | 43 | CR/CRi 61% | 17.5 mona | NCT02203773 |
| Azacitidine + venetoclax69 | 3 | ND AML | 431 | CR/CRi 66.4% | 14.7 mona | NCT02993523 | ||
| 10-day decitabine + venetoclax68 | 2 | ND AML > 60 | 168 | ORR 74% (ND AML 89%, RR AML 62%) | ND AML 18.1 mon, RR AML 7.8 mona | NCT03404193 | ||
| IDH inhibitor + BCL-2 inhibitor | Ivosidenib + venetoclax ± azacitidine83 | 1/2 | ND AML and MDS | 19 | CR 78% | NCT03471260 | ||
| Epigenetic agent + conventional chemotherapy | HDAC inhibitor + cytotoxic agents | Vorinostat + idarubicin and cytarabine86 | 2 | ND AML and MDS | 75 | CR/CRi 85% | 47 wkb | NCT00656617 |
| Panobinostat + idarubicin and cytarabine87 | 1/2 | ND AML | 38 | CR 64% | 17.0 mona | NCT00840346 | ||
| Panobinostat + azacitidine + idarubicin and cyratabine88 | 1 | ND AML and MDS | 25 | CR/CRi 32% | 23 mon with CR/CRi | |||
| 7.8 mon without CR/CRi | ||||||||
| Epigenetic agent + immune checkpoint inhibitors | Oral HMA + PD-L1 inhibitor | Guadecitabine + atezolizumab77 | 1/2 | RR MDS and chronic myelomonocytic leukemia | 9 | ORR 33% | Not reached median OS | |
| HMA + PD-1 inhibitor | Azacitidine + nivolumab78 | 2 | ND MDS | 20 | ORR 80% | Not reached median OS | ||
| Azacitidine + nivolumab79 | 2 | RR AML | 70 | ORR 33% (CR/CRi 22%) | Not reported | Grade 3–4 irAEs 11% | ||
| HMA + PD-1 inhibitor + CTLA-4 inhibitor | Azacitidine + nivolumab + ipilimumab80 | 2 | RR AML | 31 | ORR 44% (CR/CRi 36%) | 10.5 mona | ||
HMA = hypomethylating agent, HDAC = histone deacetylase, ND = newly diagnosed, AML = acute myeloid leukemia, CR = complete remission, IDH = isocitrate dehydrogenase, FLT3 = fms related receptor tyrosine kinase 3, RR = relapsed or refractory, PR = partial remission, BCL-2 = B-cell chronic lymphocytic leukemia/lymphoma 2, MDS = myelodysplastic syndrome, CRi = complete remission with incomplete platelet recovery, ORR = overall response rate, OS = overall survival, PD-1 = programmed cell-death protein-1, PD-L1 = programmed cell death protein 1 ligand-1, CTLA-4 = cytotoxic T-lymphocyte-associated protein 4, irAEs = immune-related adverse events.
aMedian overall survival; bMedian event-free survival.
HMAs and HDAC inhibitors
Even though the synergistic effects of HDAC inhibitor and HMA was presented in vitro,49 randomized trials in human with combination of HMAs and HDACs (entinostat or vorinostat) did not yield survival benefit owing to increased toxicities resulted in early discontinuation of treatment.13505152 In recent trials, pracinostat, one of an oral pan-HDAC inhibitor showed positive results. In phase II trials with untreated or HMA refractory MDS patients, pracinostat plus azacitidine did not show improved outcomes.5354 However, in newly diagnosed elderly AML, pracinostat and azacitidine combination therapy presented 19.1 months of median OS and 52% of CR rate that was favorable to the historic azacitidine data.55 Subsequent phase III trial with pracinostat in combination with azacitidine in AML was conducted, unfortunately, this trial has been terminated due to a lack of efficacy (NCT03151408).
HMAs and IDH inhibitors
As increased 2-HG by IDH mutations leads to DNA hypermethylation and frequent co-occurrence of IDH and DNMT3A mutations is reported,26 combination of IDH inhibitors and DNMT inhibitors is logical therapeutic strategy. While enasidenib and ivosidenib monotherapy have shown efficacy in AML, later phase trials of IDH inhibitors and HMAs are ongoing. In a recent phase II trial, enasidenib and azacitidine combination presented 48% of ORR and 22 months of OS with acceptable toxicities in unfit AML.56 Another IDH inhibitor, ivosidenib also has shown efficacy with azacitidine in newly diagnosed unfit AML patients. In the phase II trial, ivosidenib plus azacitidine presented 78% of ORR that exceeded those of azacitidine alone.1957 Based on these data, a phase II study with enasidenib plus azacitidine (NCT02677922) and a randomized phase III AGILE study with ivosidenib plus azacitidine (NCT03173248) are ongoing.
HMAs and tyrosine kinase inhibitors (FLT3 inhibitors)
Multikinase inhibitors midostaurin, sorafenib and more specific tyrosine kinase inhibitors quizartinib and gilteritinb have been FDA approved or under development in AML.8 FLT3 ligand expression is decreased in HMA treated patients and persistence existence of FLT3-mutated leukemic cells in BM milieu after treatment can be eradicated by combination.5859 The rationale of combination of HMAs and FLT3 inhibitors are based on those results of previous studies. In a phase I/II trial of midostaurin and azacitidine combination in AML and MDS, ORR was 26% and median remission duration was 20 weeks.60 More recent phase II study of midostaurin and azacitidine combination, clinical response was 29% and median OS was 244 days.61 Clinical trials of more specific FLT inhibitors including quizartinib or gilteritinib combination with HMAs are currently being conducted (NCT03661307, NCT01892371, and NCT02752035). In a recently published abstract, patients who had received gilteritinib and azacitidine combination showed 80% of ORR with 67% of CR in newly diagnosed AML patients ineligible for intensive chemotherapy.62 The combination of quizartinib and azacitidine also have presented promising results with 76% of CR and 13.4 months of median OS.63 Phase II trials of quizartinib plus azacitidine or cytarabine combination (NCT01892371), quizartinib decitabine and venetoclax combination (NCT03661307) are ongoing. For gilteritinib, a phase III trial combining with azacitidine in newly diagnosed AML is under recruit (NCT02752035).
HMAs and BCL-2 inhibitor
A specific BCL-2 inhibitor, venetoclax is a BCL-2 inhibitor that reverses apoptotic pathway inactivation in hematologic malignancies. Although venetoclax is not an epigenetic modulator, venetoclax monotherapy showed modest activity with 19% of CR rate in relapse refractory AML.64 However, combination with HMA in AML have yielded much promising results. Venetoclax and azacitidine combination received US FDA approval based on positive results of multiple studies.6566 In a phase I therapy combining venetoclax with decitabine or azacitidine in elderly newly diagnosed AML patients, 61% achieved CR with incomplete platelet recovery (CR/CRi).67 Recently, a phase II trial of 10-day decitabine with venetoclax in intensive chemotherapy ineligible treatment-naïve or relapsed/refractory AML presented 74% of ORR (89% in treatment-naïve, 62% in relapsed/refractory) with acceptable safety. The median OS was 18.1 months in the treatment-naïve group and 7.8 months in the relapsed/refractory group.68 Finally, in a recent phase III trial a superior OS (14.7 vs. 9.6 months) and CR/CRi rate (66.4% vs. 28.3%) of venetoclax and azacitidine combination compared to azacitidine plus placebo in treatment-naïve elderly AML were proven.69 Multiple phase III trials with venetoclax and HMA combination in other conditions are actively ongoing.
FLT3 mutation, one of the most common mutation in AML presents in only around 20% of patients. Venetoclax targeting BCL-2 protein, however, does not need mutations of BCL-2 genes. The possibility of application to more patients is one of the major benefits of venetoclax-based treatment. Prevention of tumor lysis syndrome (TLS) is recommended on venetoclax combination therapy.70 Even though the TLS event of AML has not been reported, fatal TLS events occurred in chronic lymphocytic leukemia.71 TLS is an initial clue of effective treatment with rapid degradation of cancer cells though it is a serious complication. Due to the broad applicability and promising efficacy, venetoclax combination with HMAs could be expected as one of the most important treatment strategies in AML.
HMAs and immune check point inhibitors
Ipilimumab, a cytotoxic T-lymphocyte-associated protein 4 (CTLA-4) inhibitor presented efficacy for patients with relapse of hematologic malignancies after allogeneic stem cell transplantation.72 It is reported in previous researches that epigenetic silencing of immune-related genes is important to immune modulation involving T-cells in cancer.73 T cell co-stimulatory molecule such as programmed cell death protein 1 (PD-1) expression is inversely correlated with DNA methylation and HMAs upregulate the expression of immune checkpoint molecules.87374 HMAs monotherapy is not helpful in immune-wise by hampering anti-tumor immunity, however, combination with immune checkpoint inhibitors showed different results. In non-small cell lung cancer, azacitidine pre-treatment led enhanced response to immune check point inhibitors.75
Various clinical trials are ongoing combination of HMAs and immune check point inhibitors such as nivolumab, pembrolizumab, durvalumab, atezolizumab and ipilimumab.76 Guadecitabine, a novel oral HMA did not present a promising efficacy as monotherapy, however, combination with atezolizumab, a programmed cell death protein ligand-1 (PD-L1) inhibitor, presented 33% of ORR in a phase I/II study of refractory MDS and a phase II study is ongoing.77 Azacitidine and nivolumab, a PD-1 inhibitor combination presented 80% of ORR in newly diagnosed MDS with manageable immune-related adverse events (irAEs).78 In relapsed/refractory AML, a combination of azacitidine and nivolumab presented 33% of ORR (CR/CRi 22%) with 11% of grade 3-4 irAEs.79 Many current active trials with nivolumab and HMAs presented higher response rate with manageable toxicities implying newer treatment strategies. Recently, combining azacitidine and dual blockade of immune check point with nivolumab and ipilimumab in released/refractory AML is ongoing. The ORR was 44% and median OS was 10.5 months in the azacitidine, nivolumab and ipilimumab combination group.80 Most of combination trials are still in early phases. Even the early termination of trial with azacitidine and atezolizumab combination in refractory MDS has been occurred due to unfavorable hematologic toxicities.81 However, with promising results of other trials, immune checkpoint inhibition with HMAs could be a treatment breakthrough.
IDH inhibitors and BCL-2 inhibitor with or without HMAs
IDH mutation sensitizes leukemic cells to BCL-2 inhibition by 2-HG accumulation.27 This finding implies the possible efficacy of venetoclax in IDH-mutated AML. As IDH mutated relapsed/refractory AML had a superior ORR (33% vs. 10%) to wild-type in a venetoclax monotherapy trial,64 combination studies of IDH inhibitors and venetoclax have been conducted. This combination showed superior anti-leukemic activity in patient-derived xenograft,82 and led earlier phase human trials. In a recent phase Ib/II study with high-risk MDS and AML, across all treatment groups, the overall CR rate was 78% (67% in ivosidenib plus venetoclax 400 mg, 100% in ivosidenib plus venetoclax 800 mg and 67% in ivosidenib, venetoclax 400 mg and azacitidine) with acceptable toxicities. The median time to response was 2 months. And half of the patients with CR also were negative for minimal residual disease.83 Those findings imply deeper responses and even cure might be achieved with doublet or triplet combination. Early phase trials of IDH inhibitors and venetoclax with or without azacitidine are under active recruit.
HDAC inhibitors and conventional chemotherapy
As combination of HMAs and conventional cytotoxic chemotherapy presented modest activity a decade ago,8485 HDAC inhibitors, another class of epigenetic regulators were studied in combination of intensive chemotherapy. Vorinostat plus idarubicin and cytarabine in untreated AML or high-risk MDS presented CR/CRi 85% and event-free survival 47 weeks.86 Panobinostat also presented clinical activity in combination with idarubicin and cytarabine in a phase Ib/II trial with elderly untreated AML. In that trial, CR rate was 64% and median OS was 17 months with acceptable safety.87 In a recent published study, panobinostat and azacitidine combined with intensive induction chemotherapy in older patients presented 32% of CR/CRi rate and 10 months of median OS. Though for the group without CR/CRi the median OS was only 7.8 months, for the group with CR/CRi the median OS was 23 months. In this trial, panobinostat-induced increasement in histone acetylation was significantly associated with CR/CRi.88
CONCLUSIONS
Genes associated with epigenetic mechanisms tend to be more altered in elderly than younger patients. DNMT3A, TET2, and IDH mutations present 3-5 times more in elderly AML and MDS patients than in younger ones, according to The Cancer Genome Atlas (TCGA) data. More than 50% of AML occurs in old and unfit patients. In the current consensus of AML, still the complete extinction of leukemic cells without intensive cytotoxic agent is not possible. However, we might be able to find a clue of next step in combination therapies. Research in the genomic era has elucidated the reason for the complicated nature of AML. Various types of structural chromosomal abnormalities and intra-genetic aberrations occur in AML, as well as epigenetic changes. A combination of epigenetic therapies and kinase receptor inhibitors or immune checkpoint inhibitors are under investigation. Most epigenetic agents are not as toxic as conventional chemotherapy. However, one of the challenging factors associated with epigenetics-based therapy is which subset of patients could get most benefits. Among epigenetic regulators completed development and under development, except IDH inhibitors, most of agents does not have biomarkers predicting prognosis. In a recent trial with panobinostat, panobinostat-induced increase of histone acetylation was regarded as a predictor of response,88 however, more general factors to apply all patients and more earlier predictors to forecast before treatment initiation are needed. For a more effective and safe use of drugs in frail patients, biomarkers predicting drug response and adverse events must be investigated.
The relationship between the leukemogenic process and epigenetic changes remain incompletely elucidated. However, it remains clear that to multi-target as opposed to single-target strategies are required. Despite being incompletely elucidated, epigenetic therapies for AML might be one of the most important future treatment options, with present rapid development that correspond to clinical requirements.
 XML Download
XML Download