

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Gallstones in the gallbladder or bile ducts can lead to serious complications. However, the current principle of treatment of gallstones is surgical cholecystectomy upon the development of complications, or presentation of marked colic. The same principle of treatment is applied to the high risk group of gallstones.1 This might be because the cholecystectomy is regarded as a simple operation through laparoscopy, which takes a light look at the fatal situations. On the other hand, it is also true that the medical treatments have not been proved to be satisfactory. Moreover, there are few studies on pathological and biochemical mechanisms contributing to medical treatment or prevention.
The cholesterol saturation index (CSI) in bile, determined by the composition of cholesterol, bile acids, and phospholipids, has a pivotal role in the maintenance of solubility of cholesterol.2 Nuclear hormone receptors including farnesoid X receptor (FXR), peroxisome proliferator-activated receptor α and γ (PPARα, PPARγ), and liver X receptor α (LXRα), are related to cholesterol and bile acid metabolism, and affecting each other, in the nuclei of hepatocytes.3-5 Previous our animal study has proved that pravastatin activates PPARγ in the liver and gallbladder epithelium, and suppresses the formation of cholesterol stones or crystals in the gallbladder.6 Since then, we have tried to reveal the transcription mechanism that statin prevents of cholesterol gallstone formation.
FXR is involved in maintaining the proper concentration of bile acid in the blood.7-9 A study using a mouse model has demonstrated that the synthesized FXR ligand prevented the formation of cholesterol gallstones.10 Currently, we do not have suitable agonists that directly stimulate FXR without an increase in the concentration of bile acid in the blood. Recently, and we have demonstrated that statin, used for the management of hypercholesterolemia, increased the expression of FXR.11 Similarly, we aimed to demonstrate the possible role of statins as FXR agonists.
Statin is suggested to activate PPAR coactivator-1α (PGC-1α) by inhibiting Akt phosphorylation, activating FXR as a result of interaction with HNF-4α which attaches to the DR-1 element of the PPARγ and FXR promoters.12-14 However, this speculation is debatable, and it is still unclear how PGC-1α, PPARγ and HNF-4α affect each other. On the other hand, we have also observed that statin increased the CYP7A1 mRNA expression via the PPARγ and LXRα pathways, although there is no LXR response element (LXRE) in the CYP7A1 promotor of human hepatocyte and activated FXR suppresses CYP7A1.11,15-17 Given that statin induces PPARγ, and PPARγ activation increases CYP7A1 mRNA expression, it is presumed that the statin is involved in PPARγ/PGC-1α/HNF-4α pathway, and induces activation of CYP7A1. However, this has not yet been verified.
Therefore, this study was performed to understand how PPARγ, PGC-1α and HNF-4α are involved in the statin-induced activation of FXR and CYP7A1, how they affect each other, and also to verify whether the mevalonate pathway is involved in this mechanism.
SUBJECTS AND METHODS
1. Materials
Fetal bovine serum (FBS), Dulbecco's modified eagle medium (DMEM), trypsin/EDTA and penicillin/streptomycin were purchased from Gibco (Grand Island, NY, USA). Atorvastatin was procured from Sigma (St. Louis, MO, USA). WY14643 and troglitazone were procured from Cayman Chemicals (Ann Arbor, MI, USA). The rabbit polyclonal antibodies for PPARγ, FXR, CYP7A1, PGC-1α and HNF-4α were purchased from Abcam (Cambridge, UK). Goat anti-rabbit IgG-HRP and the β-actin antibody were procured from Santa Cruz Biotechnology (Santa Cruz, CA, USA), and the Amersham ECL™ Advance Western Blotting Detection Kit was purchased from Amersham Biosciences (Buckinghamshire, UK).
2. Cell culture
Human Hep3B cells, harvested from hepatocellular carcinoma, were purchased from the Korean Cell Line Bank (KCLB). The absence of mycoplasma or bacteria contamination and the cell line short tandem repeat (STR) profile was authenticated by KCLB. We cultured the cells in DMEM with 4.5 g/L glucose and 10% FBS, supplemented with 1.5 g/L sodium bicarbonate, 2 mM glutamine, 100 IU/mL penicillin, and 100 μg/mL streptomycin. The media was changed 2 times a week and cells were incubated at 37℃ in a humidified incubator with 5% CO2. Cells were passaged when cultures attained confluency (every 5-7 days) using trypsin (2.5 g/L) and EDTA (1 g/L).
3. MTT (3-[4,5-dimethylthiazol-2-yl]-diphenyltetrazolium bromide) assay
Cell proliferation was measured using the MTT assay. Cells were plated at a density of 5×10⁴ cells/mL in regular culture media in 96-well plates for 24 hours. The cells were loaded with indicated concentrations of atorvastatin in serum-free medium (SFM) for 24 or 48 hours. Each well was added with MTT (0.5 mg/mL) (Sigma, St. Louis, MO, USA), and then incubated longer for 4 hours at 37℃. Following removing the culture media, 100 μL of DMSO was treated to each well. The colorimetric determination was evaluated using DTX 880 Multimode Detector (Beckman Coulter, Brea, CA, USA) at 570 nm.
4. Western Blotting Assay
Harvested cells after washing with PBS were liquefied using lysis buffer (pH 7.5-50 mM Tris, 1% Tripton X-100, 150 mM NaCl, 1 mM EDTA, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 μM phenylmethylsulfonyl fluoride (PMSF), 5 μg/mL leupeptin, 5 μg/mL aprotinin). The protein content of the cell lysate was estimated using the Bradford assay (Sigma, St Louis, MO, USA). SDS--polyacrylamide gel electrophoresis was performed with indicated stacking and resolving gel, and then transferred to a nitrocellulose membrane (Bio-rad, Hercules, CA, USA). Blots were blocked in blocking solution (5% skimmed milk in pH 7.5 Tris-buffer with Tween-20 [TBS-T]: 200 mM Tris, 500 mM NaCl, 0.05% v/v Tween-20) at room temperature, and then incubated overnight at 4℃ in 5% bovine serum albumin in TBS-T with the rabbit polyclonal antibodies for PPARγ, FXR, CYP7A1, PGC-1α, and HNF-4α. The membranes were rinsed using TBS-T and incubated with goat anti-rabbit IgG-HRP for 1 hours at room temperature. The Blots were washed and treated with reagents from the Amersham ECL™ Advance Western Blotting Detection Kit for 2 min, and autoradiography was obtained. Image J density analysis software (National Institute of Health, Version 1.43) was used to quantify signal intensities for specific bands on the blots.
5. Transfection of siRNA
Hep3B cells were placed at a density of 5×105 cells/well, grown to confluency of 40-50%, and transfected with siRNA oligonucleotides that targeted PPARγ, PGC-1α, HNF-4α and non-silencing control siRNA (Invitrogen, Carlsbad, CA, USA). Every transfection required 20 pmol siRNA oligomer in 500 μL serum-free OPTI MEM with 3 μL lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA). The cells were transfected in 1.5 mL of growth medium without antibiotics for 48 hours, then rinsed, and incubated with SFM containing 1% BSA (Sigma, St Louis, MO, USA) for 24 hours. The efficiency of siRNA transfection was assessed using Western blotting.
6. Statistical analysis
All the described results are representative data of three separate experiments at least, and expressed as the means ± SD of duplicate cultures. The Student’s t-test was used for two unpaired groups, and the one-way ANOVA test was used for three or more unpaired groups. P-value below 0.05 was considered statistically significant.
RESULTS
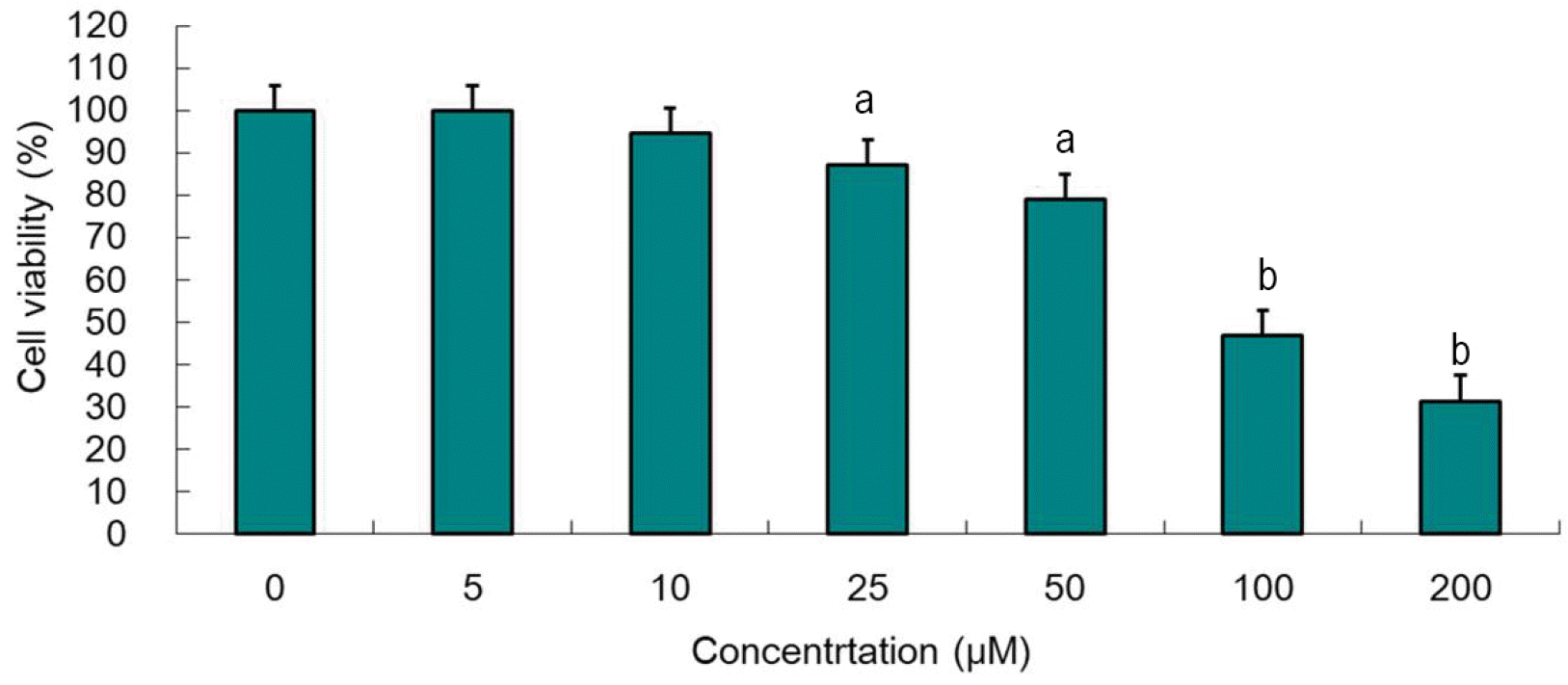
1. Atorvastatin suppresses the proliferation of Hep3B cells
Atorvastatin-induced inhibition of Hep3B cell proliferation was measured by the MTT assay after 24 hours incubation. Atorvastatin suppressed Hep3B cell proliferation dosedependently. Concentrations over 25 μM significantly affected cell growth in 24 hours (Fig. 1). We then determined the loading dose of atorvastatin to 10 μM or less for Western blotting to show changes in the expression of proteins.
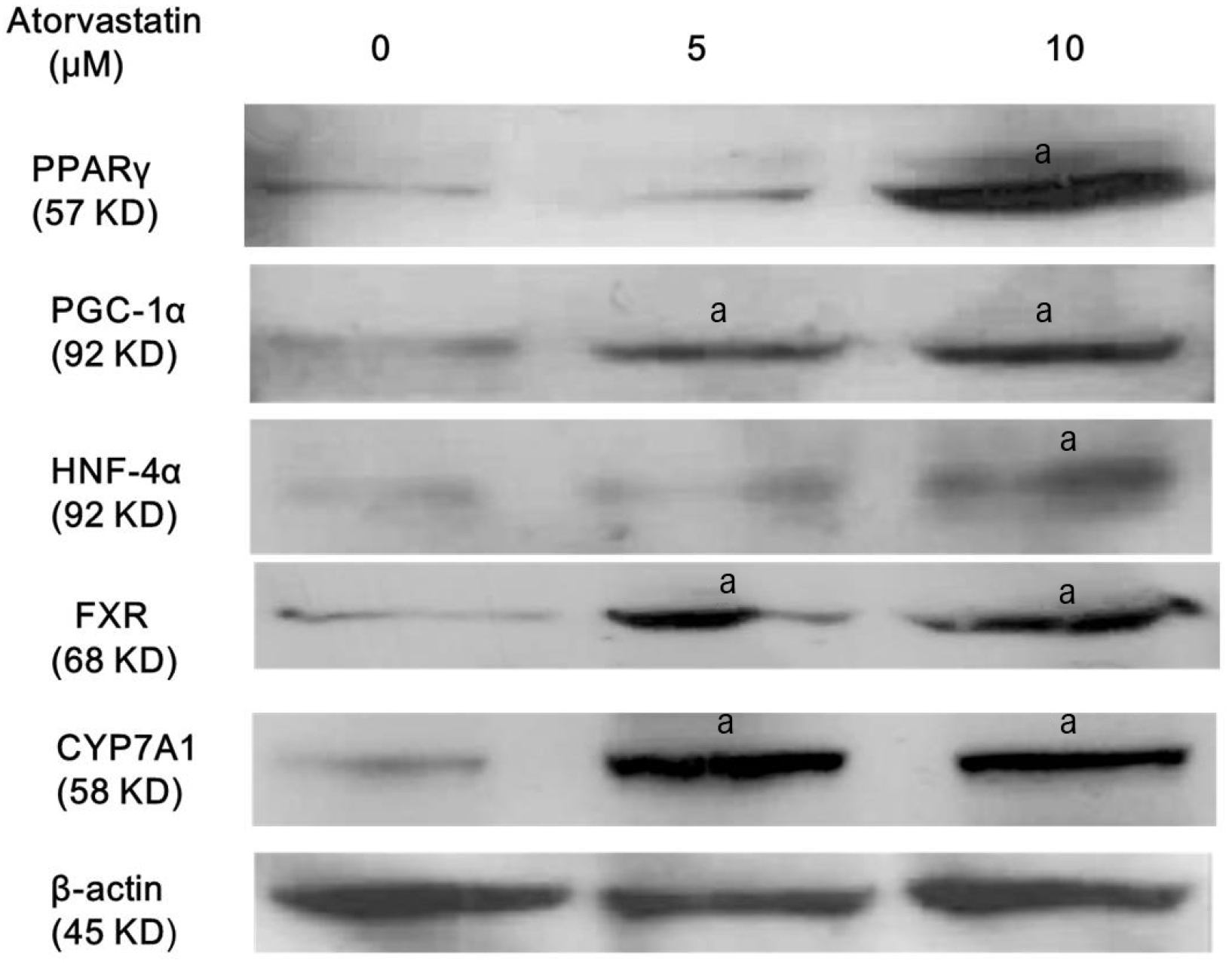
2. Atorvastatin increases expression of PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1 in Hep3B cells
To evaluate whether atorvastatin affects expression of PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1, Western blotting was performed for PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1 after loading of scheduled concentrations of atorvastatin for 24 hours. Atorvastatin induced activation of PPARγ, PGC-1α, HNF-4α, and CYP7A1 in a dose-dependent or independent manner (Fig. 2).
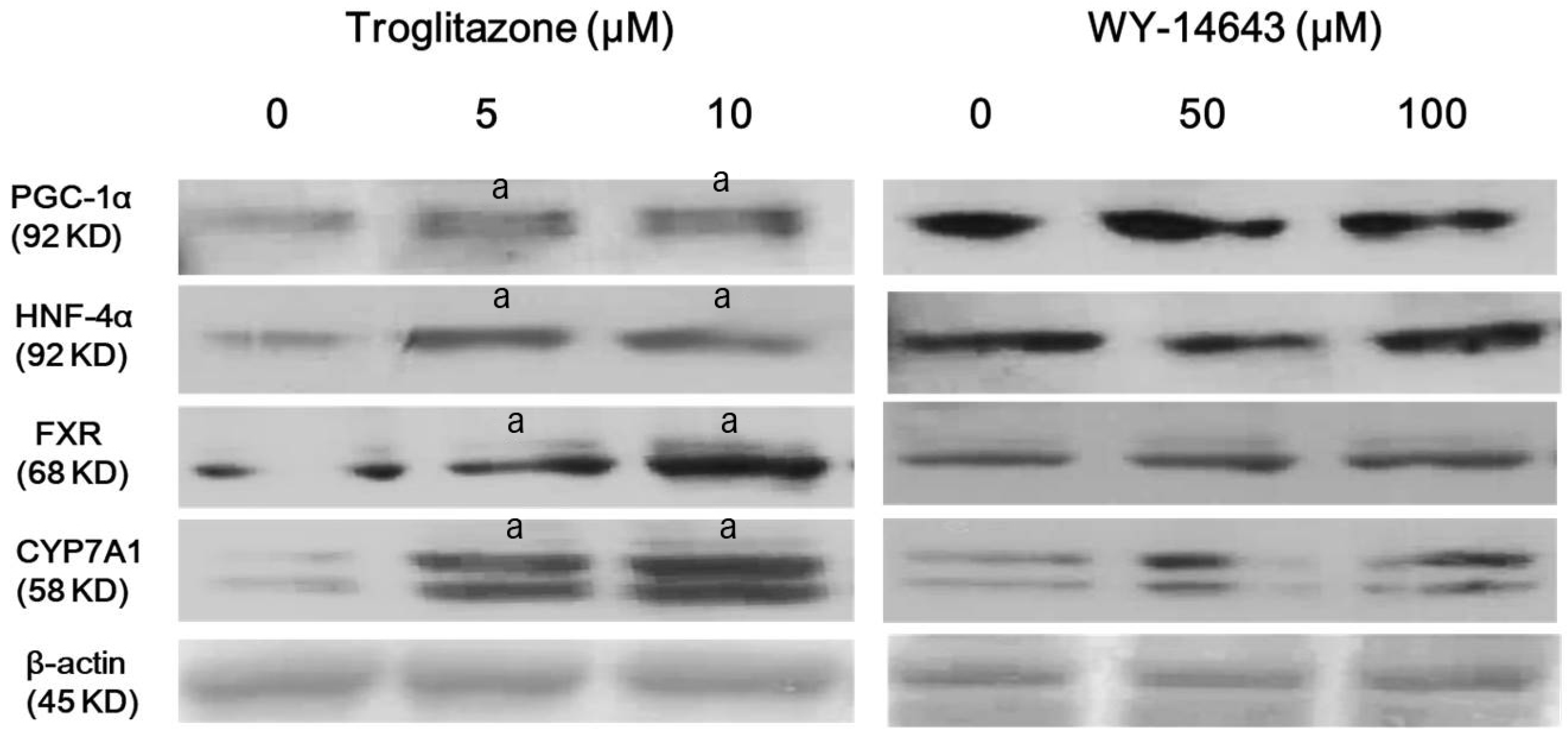
3. PPARγ ligand increases expression of PGC-1α, HNF-4 α, FXR, and CYP7A1 in Hep3B cells
Western blotting was performed for PGC-1α, HNF-4α, FXR and CYP7A1 after treatment of indicated concentrations of troglitazone (PPARγ ligand) or WY-14643 (PPARα ligand) for 24 hours. Troglitazone induced activation of PGC-1α, HNF-4α, FXR, and CyP7A1. However, WY-14643 did not induce a significant change in expression of PGC-1α, HNF-4α, FXR and CYP7A1 (Fig. 3). These findings support that PPARγ is mainly involved in activating HNF-4α, FXR and CYP7A1 as compared to PPARα.
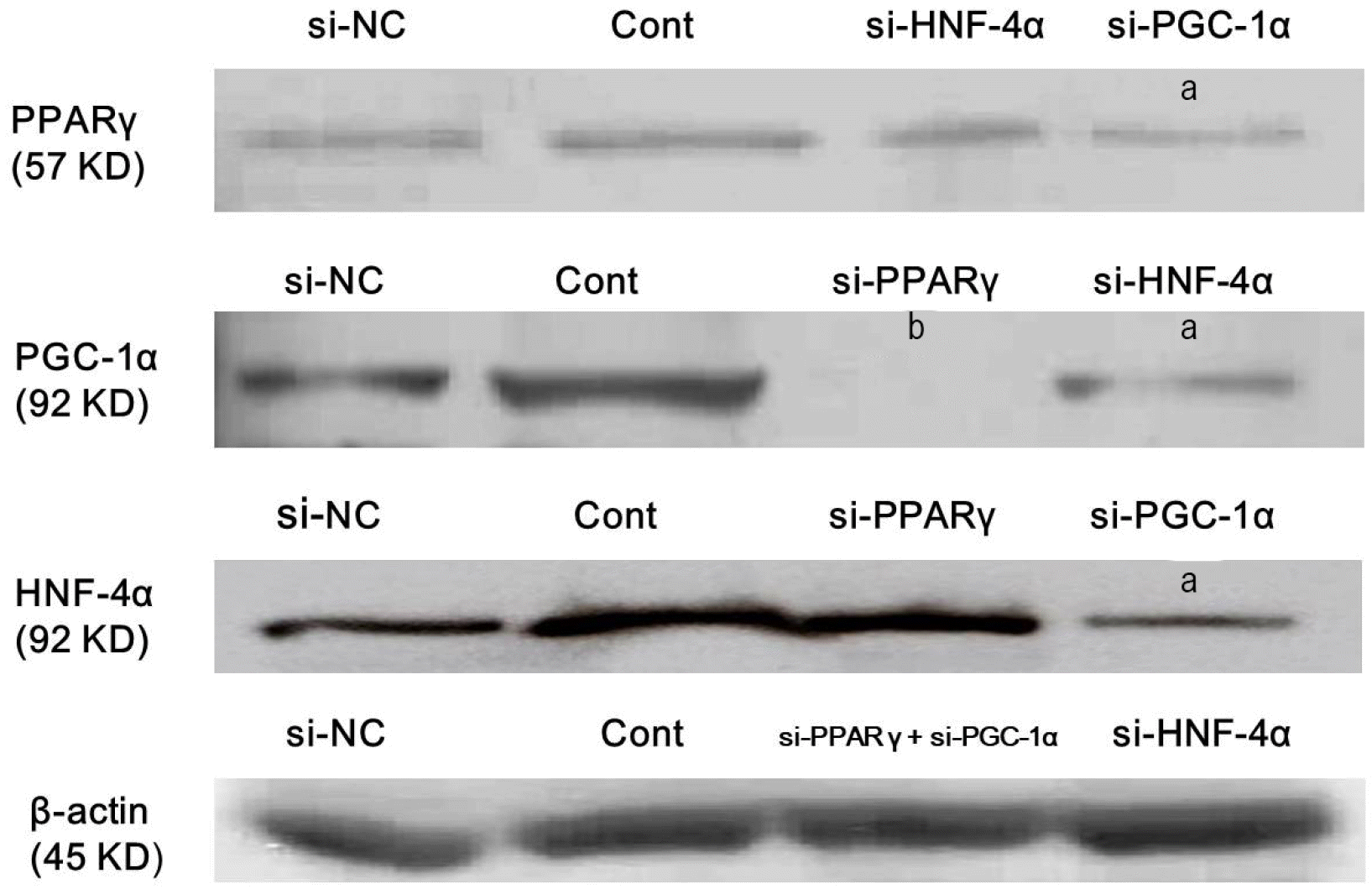
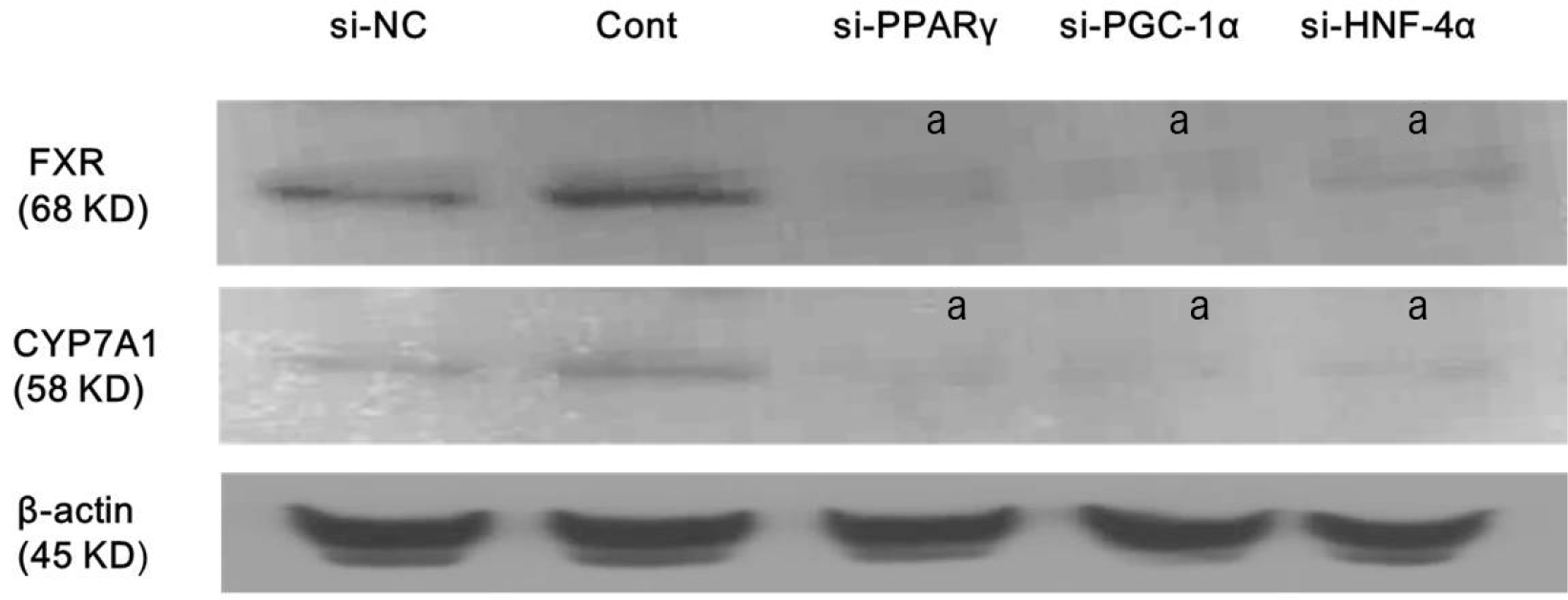
4. Atorvastatin-induced FXR and CYP7A1 activation requires sequential action of PPARγ/PGC-1α/HNF-4α
Upon treatment with or without 10 μM atorvastatin for 24 hours, expression of PPARγ, PGC-1α, HNF-4α, FXR and CYP7A1 was evaluated following siRNA transfection against PPARγ, PGC-1α, or HNF-4α to identify the key protein involved in activation of these specific proteins. Change in expression of PPARγ after siRNA transfection for PGC-1α or HNF-4α showed that PGC-1α silencing partially inhibited atorvastatin-induced PPAR γ expression, and HNF-4α silencing did not affect atorvastatin-induced PPARγ expression. Interestingly, PPARγ silencing completely inhibited atorvastatin-induced PGC-1α expression, and HNF-4α silencing partially inhibited atorvastatin-induced PGC-1α expression. These results show that PGC-1α is necessary for complete PPARγ activation, whereas PPARγ activation is essential for the activation of PGC-1α. Change in expression of HNF-4α after siRNA transfection against PPARγ or PGC-1α indicated that HNF-4α activation is dependent on PGC-1α rather than on PPARγ (Fig. 4).
Silencing of PPARγ, PGC1α, and HNF4α using respective siRNA inhibits FXR and CYP7A1 expression, which means that atorvastatin-induced FXR and CYP7A1 activation requires the sequential action of PPARγ/PGC-1α/HNF-4α or interactions among them (Fig. 5).
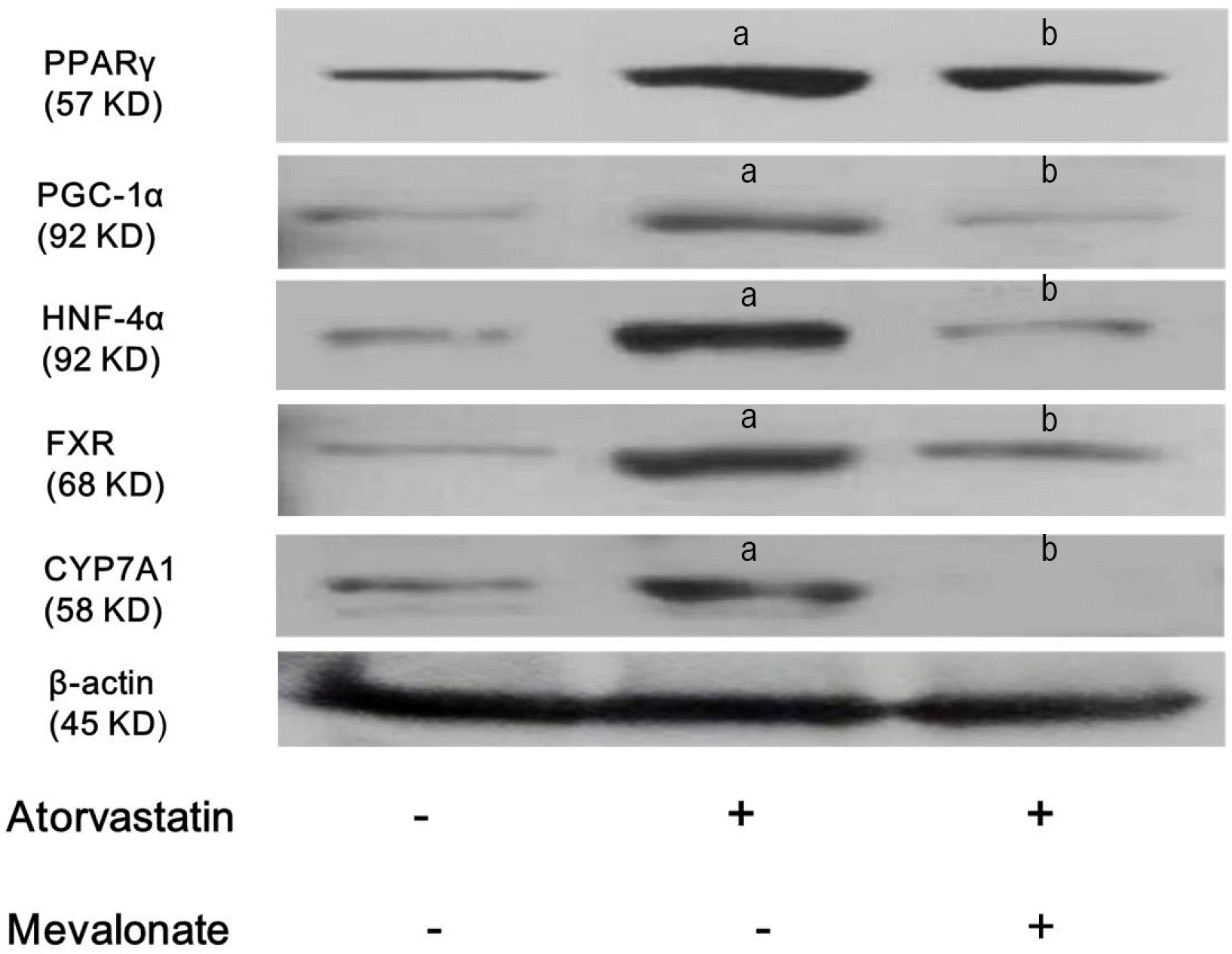
5. Atorvastatin-induced suppression of the mevalonate-isoprenoid pathway is related to activation of PPARγ, PGC-1α HNF-4α, FXR, and CYP7A1
To determine whether the mechanism of action of atorvastatin in increasing expression of PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1 is associated with the mevalonate–isoprenoid pathway, Western blotting was performed for PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1 after 10 μm atorvastatin treatment for 24 hours, with or without 100 mM mevalonate pre-treatment for 2 hours. Mevalonate completely reversed the effect of atorvastatin on PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1 (Fig. 6), which means that statin-induced inhibition of the mevalonate– isoprenoid pathway via the 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase block is linked with the activation of PPARγ, PGC-1α HNF-4α, FXR, and CYP7A1.
DISCUSSION
This study is significant for the following reasons: 1) we have proved that statin activates FXR and CYP7A1, and have shown the mechanisms involved as well, 2) the mevalonate pathway is clearly associated with the signal translation by which statin increases expression of PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1, and 3) we have demonstrated that PGC-1α has a crucial role in HNF-4α activation, and PGC-1α is necessary for PPARγ activation, whereas, PPARγ is essential for activation of PGC-1α.
The primary target of the MTT assay was to determine an adequate loading dose of atorvastatin for Western blotting to show changes in expression of proteins of Hep3B cells, revealing that atorvastatin suppresses Hep3B cell proliferation dose-dependently (Fig. 1). This result showed a possibility of direct toxic or anti-neoplastic effect of atorvastatin on hepatoma cells. Our previous study using extrahepatic bile duct cancer cells has proved that statins have anti-neoplastic effect.18 However, there has been a report that statins have direct toxicity to rat hepatocytes.19 Accordingly, additional studies on antineoplastic effect of statins using hepatoma cell lines will be anticipated in the near future.
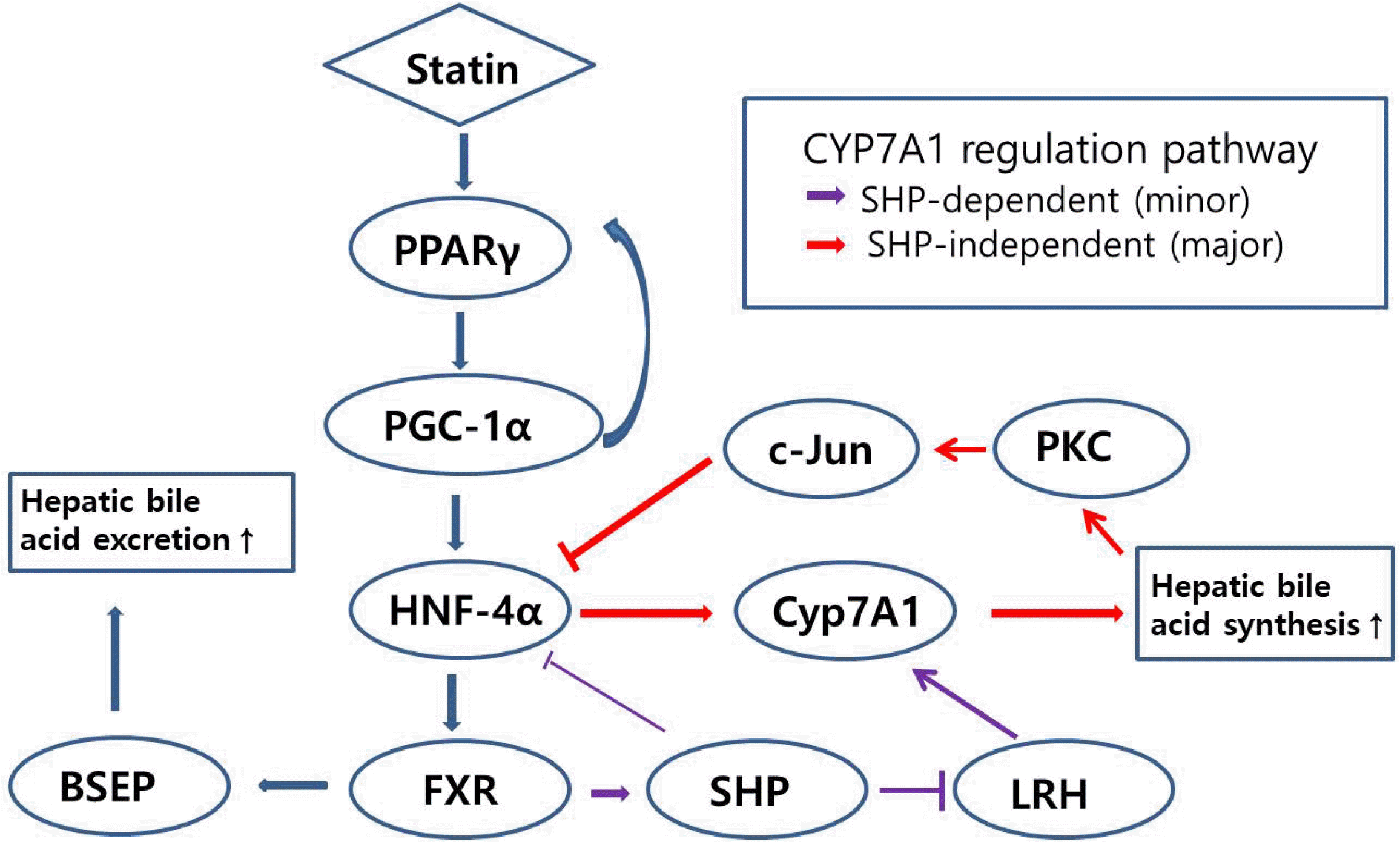
Hepatocytes synthesize bile acid from cholesterol. When the concentration of bile acid in the blood or hepatocytes increases, FXR presented in the hepatocytes is activated and inhibits hepatic 7α-hydroxylase (CYP7A1). Activated FXR acts on the salt exporter pump (ABCB11, BSEP) which excretes bile acids into bile canaliculi, and also acts on multidrug resistance gene 3 (ABCB4, MDR3,) which excretes phospholipids into bile canaliculi, resulting in lowered CSI in hepatic bile.20 The rate-limiting enzyme in the neutral pathway is CYP7A1.21 The CYP7A1 gene is known to be regulated by the small heterodimer partner (SHP)-dependent pathway which is regulated by FXR that has a site for binding of bile acids. The heterodimer, composed of FXR and RXRα, then activates SHP. The SHP binds to liver homologue receptor-1 (LRH-1), inducing its inactivation, and thereby inhibits the expression of CYP7A1. Statins were found to increase the expression of CYP7A1 in our previous11 and current studies, although statins activate FXR. Therefore, another pathway is likely to be involved in the statin-induced increase in expression of CYP7A1. Bile acids synthesis is also inhibited via an SHP-independent pathway. Bile acids stimulate protein kinase C which deactivates HNF-4α via the c-Jun kinase pathway, resulting in failure of HNF-4α-induced CYP7A1 gene activation. Besides, SHP is able to inactivate HNF-4α.21,22 This hypothesis was supported by some studies. One study showed that expression of HNF-4α has a positive correlation with one of CYP7A1, and that chenodeoxycholic acid (CDCA) treatment to hepatocytes decreased the expression of HNF-4α. The study concluded that the positive feedback of CYP7A1 by HNF-4, the SHP-independent pathway, is predominant compared with the SHP-dependent pathway.23 Another study has also reported that CDCA moderately suppressed the activity of the human HNF-4α promoter in cultured HepG2 cells.24 We observed that atorvastatin induced activation of HNF-4α and CYP7A1 despite activation of FXR, which was in accordance with the conclusion of the study of Abrahamsson et al.23 Besides, HNF-4α silencing using siRNA did not show activation of CYP7A1 upon treatment with atorvastatin, which means HNF-4α has a pivotal role in the activation of CYP7A1. Suggestive mechanism of statin that activates and regulates FXR and Cyp7A1 in terms of bile acid metabolism in hepatocytes is summarized as a schematic illustration in Fig. 7.
HNF-4α exists in the liver, kidney, intestine, and pancreas,25 and plays a crucial role in lipid and glucose metabolism in the liver.26,27 PGC-1α, transcriptional coactivator, controls processes of energy utilization and production.28 Fasting, in the liver, induces activation of PGC-1α, stimulating gluconeogenesis, at least partially by stimulatory effect of PGC-1α on HNF-4α.29 Fasting is also known to change the expression of bile acid transporters through PGC-1α and HNF-4α in hepatocytes.30 Our data showed that PGC-1α silencing upon atorvastatin stimulation failed to induce expression of HNF-4 α, which means that PGC-1α is an essential factor required to activate HNF-4α. A study has demonstrated that cAMP-PKA enhances HNF-4α expression as a result of PGC-1α-induced CREB activation.31
There have been several debates on whether or not statins activate the expression of FXR. One study using the hamster model and HepG2 cells observed that simvastatin decreased expression of FXR.32 Another study supported the hypothesis by demonstrating that atorvastatin suppressed SHP and fibroblast growth factor 15 which are target genes of FXR. The study also suggested that atorvastatin-induced CYP7A1 activation is a result of inhibited FXR signaling in the liver and intestine.33 However, a study has examined whether of statins activate expression of FXR using nuclear receptor activation assay, which showed that simvastatin, fluvastatin, rosuvastatin and pravastatin activate FXR dose-dependently.34 These results were also repeated in our previous study which demonstrated that pravastatin suppressed cholesterol gallstone formation through increased activity of FXR, LXRα and CYP7A1 in human hepatocytes.11 Our study not only demonstrated evidence that statin activates FXR expression, but also revealed the mechanism that atorvastatin induces FXR activation as a result of sequential actions of PPARγ/PGC-1α/HNF-4α.
CDCA and obeticholic acid (OCA; 6a-ethylchenodeoxycholic acid) are FXR agonists which are used in clinical practice. However, there is a risk of increased bile acid concentration in blood because they belong to a class of bile acid derivatives. Other experimental FXR agonists are still being experimentally examined.35 We believe that statins can be potent FXR agonists, considering that statin had an effect on activation of FXR in the present study. We can also develop novel derivatives based on the chemical structure of statins.
In 1998, Puigserver et al.36 isolated PGC-1α, described its domains that mediate the PGC-1α/PPARγ interaction, and demonstrated that PGC-1α is a potent transcriptional coactivator for PPARγ. Also, by mapping the domain of PPARγ that interact with PGC-1α, they suggested that PPARγ utilizes part of its DNA-binding and hinge domains to bind to PGC-1α. Our study demonstrated the relationship or mutual interaction between PGC-1α/PPARγ: PGC-1α is necessary for complete PPARγ activation, and PPARγ is essential for the activation of PGC-1α.
HMG-CoA reductase, the rate-limiting enzyme of the mevalonic acid synthesis from HMG-CoA in smooth endoplasmic reticulum of hepatocyte, can be inhibited by statins.37 Prenylation of small GTP-binding protein is inhibited due to blocked synthesis of farnesyl pyrophosphate and geranylgeranyl pyrophosphate by statins along the mevalonate pathway.38 Our study showed that mevalonate completely reversed the effect of atorvastatin on PPARγ, PGC-1α, HNF-4α, FXR, and CYP7A1, which means that inhibition of the mevalonate-isoprenoid pathway as a result of statin-induced the HMG-CoA reductase inhibition is strongly associated with activation of PPARγ, PGC-1α HNF-4α, FXR, and CYP7A1.
Hep3B cells were used in the current study due to technical advantages of hepatocyte culture. Hep3B cell lines have physiologic and biochemical characteristics similar to normal primary hepatocytes. A lot of studies related to bile acid and cholesterol metabolism on transporters, enzymes, receptors in hepatocytes, such as CYP7A1,39-41 LXRα,42,43 FXR,32 and PPAR,44 have used Hep3B or HepG2 cells. However, there might be the possibility of physiological differences between Hep3B cells and primary hepatocytes, which might be a limitation of our study.
In conclusions, atorvastatin induces FXR and CYP7A1 activation as a result of sequential action of PPARγ/PGC-1α /HNF-4α, and PGC-1α is necessary for complete PPARγ activation whereas PPARγ activation is essential for the activation of PGC-1α in human hepatocytes. HNF-4α activation is dependent on PGC-1α rather than PPARγ. We suggest that statin may prevent cholesterol gallstone formation by activation of FXR increasing hepatic bile acid excretion and CYP7A1 increasing hepatic bile acid synthesis, and can be an effective FXR agonist.
 XML Download
XML Download