INTRODUCTION
Acute myeloid leukemia (AML) is a heterogeneous hematological malignancy that involves the uncontrolled clonal proliferation of blasts in the bone marrow and peripheral blood. It is characterized by clonal evolution and genetic heterogeneity [
1-
3]. Since cytogenetic profiles have become important indicators of prognosis in AML, the World Health Organization, National Comprehensive Cancer Network, and European LeukemiaNet have incorporated certain cytogenetic and molecular abnormalities in AML classifications and risk stratifications. Internal tandem duplication in FMS-like tyrosine kinase 3 (
FLT3-ITD) mutations have been categorized as poor prognostic factors. FLT3 gene mutations occur in about 30% of adult patients diagnosed with AML, but in pediatric patients, it is rare and occurs at a rate of 5–15% [
1].
FLT3-ITD triggers the activation of tyrosine kinase receptors and downstream signaling, leading to increased leukemic stem and progenitor cell proliferation and their increased survival [
4]. These patients have leukocytosis, a higher percentage of blasts, a higher relapse rate, and poorer overall survival (OS) compared to patients with wild-type (wt) [
5,
6]. Although there are no significant differences in complete remission (CR) rates, responses are usually short-lived and are inferior to salvage therapies.
FLT3-ITD mutations in the pediatric AML population have also been reported to have poorer OS and event-free survival (EFS) compared to wt [
6,
7]. A previous study from our institution in 2004 reported an OS and EFS rate of 0% in this population [
6]. A recent study reported 5-year OS and EFS rates of 42.2% and 36.8%, respectively [
8]. Due to considerable evidence of the poor prognosis associated with
FLT3-ITD mutations, many institutions have incorporated allogeneic hematopoietic stem cell transplantation (HSCT) and newer treatment options, such as FLT3-targeted therapy, into their therapeutic regimens, especially in the adult population [
5]. Nevertheless, optimal treatment has not been well-defined in the pediatric population. Thus, this retrospective study aimed to analyze the outcomes of pediatric AML patients with
FLT3-ITD mutations receiving the current treatment regimen at a single institution over a decade. This is a follow-up study from a previous report published in 2004 and is the first to focus on pediatric AML patients with
FLT3-ITD mutations in Korea. This study aims to provide baseline data for comparing the outcomes of newly incorporated target therapies.
Go to :

MATERIALS AND METHODS
Patients
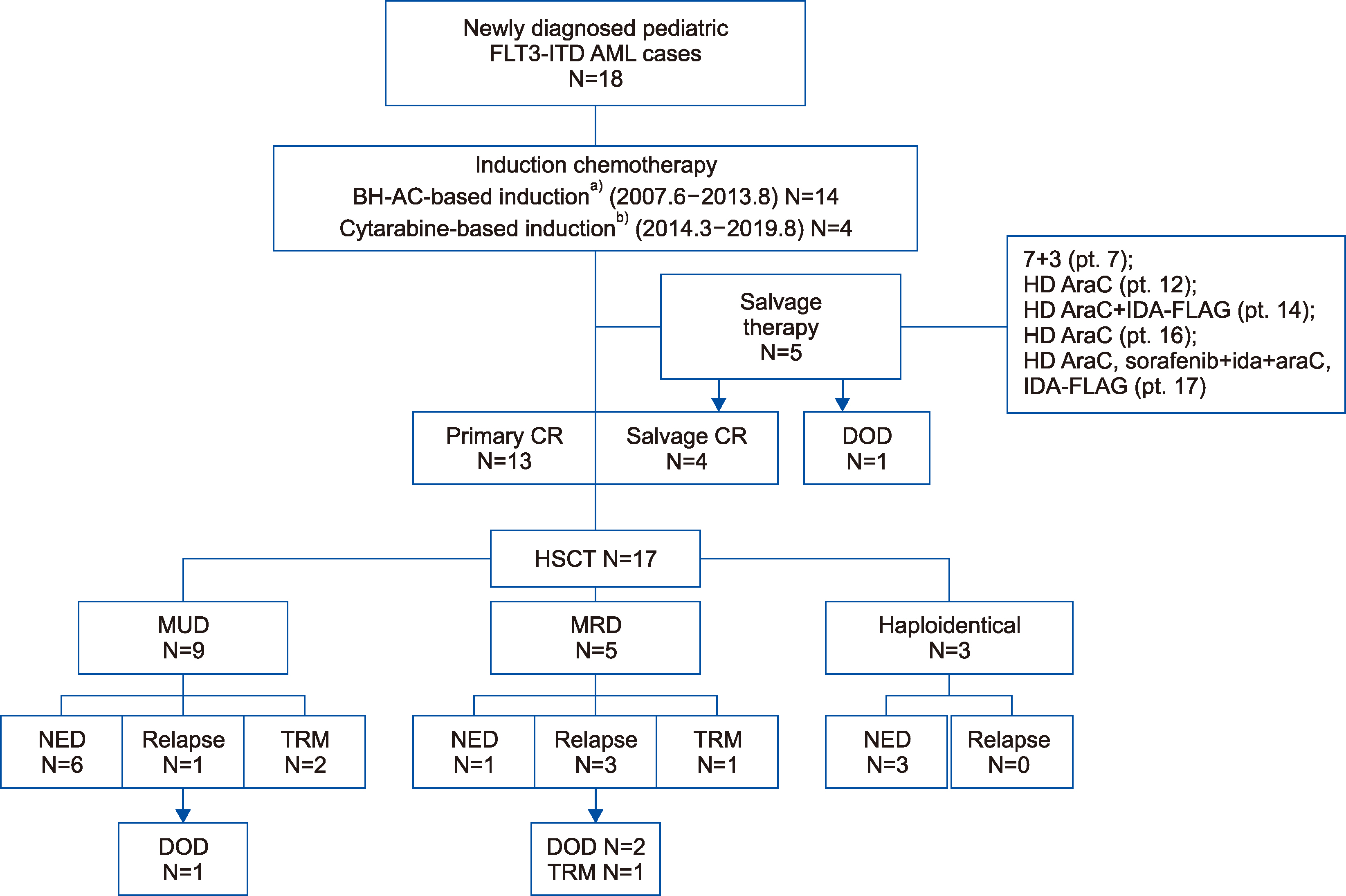
All AML patients with FLT3-ITD mutations aged 19 years and under, newly diagnosed between May 2008 and August 2019 at Seoul National University Children’s Hospital, were included in this study. Patients with acute promyelocytic leukemia were excluded. A total of 18 patients were identified for this study by a retrospective medical record review and followed until June 1, 2020. This was a retrospective, single-center study carried out after approval and waived informed consent was received from the Institutional Review Board of Seoul National University Hospital (IRB H-2004- 146-1118).
Data collection and method
Demographic characteristics, laboratory findings, treatments, toxicities, and outcome data were collected by reviewing medical records. AML classification was determined according to the French-American-British (FAB) criteria. Cytogenetic and genetic analyses were conducted using the trypsin-Giemsa banding technique and fluorescent in situ hybridization (FISH) on bone marrow cells at diagnosis. All patients had their FLT3-ITD mutation status determined by fragment analysis, polymerase chain reaction, and direct sequencing from bone marrow aspirate at diagnosis. From the initial bone marrow sample, all the patients were tested for concurrent gene mutations, including AML1/ETO (8;21), MLL rearrangement, PML-RARA, and CBFB (INV16), using FISH.
Chemotherapy regimen
Prior to 2014, the induction chemotherapy protocol for pediatric AML patients in our institution was BH-AC-based (N4-Behenoy1-1-b-D-arabinofuranosy1cytosine) and consisted of enocitabine (300 mg/m
2/day) for 7–10 days, idarubicin (IDA, 12 mg/m
2/day) for 3 days, and intrathecal (IT) cytarabine [
6]. Starting in 2014, our center began to utilize a cytarabine-based induction regimen consisting of 2 courses, the first of which involved a continuous intravenous (IV) cytarabine infusion (200 mg/m
2/day) for 7 days, IDA (12 mg/m
2/day) for 3 days, and IT cytarabine. The second course consisted of 8 doses of IV cytarabine (1,500 mg/m
2), mitoxantrone (12 mg/m
2) for 2 days, and IT cytarabine. Patients with primary refractory disease received various salvage chemotherapies, as further elucidated in the results section. After chemotherapy, patients with
FLT3-ITD mutations received high-dose conditioning therapy and allogeneic HSCT. For donor selection, human leukocyte antigen (HLA) compatibility (HLA-A, -B, -C, -DRB1, or -DQB1) was tested. Three different conditioning regimens were used in this study. One of the earlier regimens consisted of total body irradiation (TBI) at 300–333 cGy for 3 days, cytarabine (3 g/m
2) twice a day for 2 days, fludarabine (50 mg/m
2) for 4 days with or without thymoglobulin (1.5 mg/kg) for 3 days (TBIAcFluda). Another conditioning regimen consisted of targeted busulfan for 4 days, fludarabine (40 mg/m
2) for 6 days, etoposide (20 mg/kg) for 3 days, and thymoglobulin (2.5 mg/kg) for 3 days (BuFluVPATG). Depending on the type of donor (unrelated versus related), methotrexate and tacrolimus or cyclosporine were also administered for graft-versus-host disease (GVHD) prophylaxis. Lastly, the regimen used for haploidentical donors consisted of targeted busulfan for 4 days, fludarabine (40 mg/m
2) for 5 days, and cyclophosphamide at a dose of 14.5 mg/kg for the 2 days prior to the infusion and 50 mg/kg for 2 days after the infusion (BuFludaCy). For GVHD prophylaxis, mycophenolate mofetil and tacrolimus were administered. Targeted IV busulfan was administered at a starting dose of 120 mg/m
2, and subsequent doses were analyzed using daily therapeutic drug monitoring [
9-
11].
Definition of outcomes
OS was defined as the duration from diagnosis to death or last follow-up, and EFS was defined as the duration from diagnosis to the first event (consisting of death or relapse). Primary refractory disease was not considered an event. Relapse free survival (RFS) was defined as the duration in months from the end of the primary treatment (HSCT) to relapse. The occurrence of death without relapse was not included as an event for RFS. Data on living patients and those who had died from any cause without relapse were recorded until the date of death or last follow-up, using a cut-off date of June 1, 2020. CR was defined as morphological remission (<5% blasts) in the bone marrow, and primary CR (CR1) was defined as morphological remission after the first course of induction chemotherapy. Primary refractory cancer to BH-AC-based induction was defined as morphological persistence (>5% blasts) in the bone marrow after induction chemotherapy. For cytarabine-based therapy, primary refractory disease was defined as >20% blasts in the bone marrow after the first course of induction chemotherapy and/or >5% blasts in the marrow after both courses of induction chemotherapy. The neutrophil engraftment date was defined as 3 consecutive days with an absolute neutrophil count (ANC) greater than 0.5×109/L after infusion.
Statistical analysis
Clinical and laboratory data were analyzed using standard statistical methods. The OS, EFS, and RFS were analyzed using the Kaplan-Meier method and the difference in survival rates was determined using the log-rank test, with results expressed as percentages±standard error. Statistical significance was defined as a P-value <0.05. Data and statistical analyses were performed using STATA ver 13 (StataCorp LLC, College Station, TX, USA).
Go to :
DISCUSSION
This retrospective study outlined the clinical course and evaluated the outcomes of pediatric AML patients with
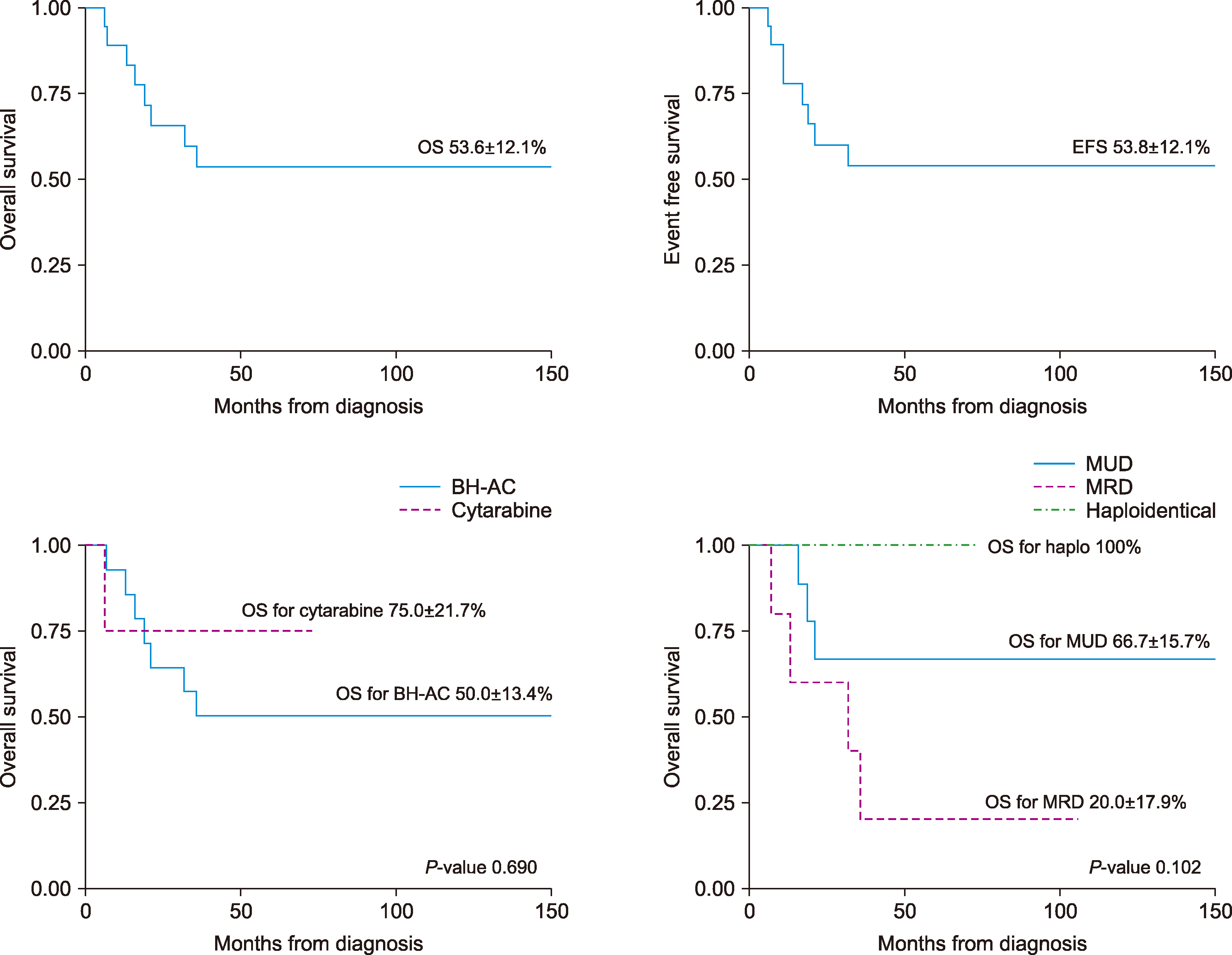
FLT3-ITD mutations in a single institution over a period of more than 20 years. However, during these 2 decades, many changes in treatment occurred. The induction regimen changed from BH-AC-based to cytarabine-based chemotherapy, the conditioning chemotherapy regimen for HSCT was altered slightly over time, and, recently, haploidentical HSCT has become more common. However, there were no significant differences in survival outcomes based on the induction chemotherapy regimen or HSCT donor type (
Fig. 2). Currently, in our institution, the two preferred donor types are the MRD and the MUD. However, to avoid delays when searching for a matched donor, haploidentical donors are becoming more common. Previously, a higher incidence of TRM caused by HLA mismatches was found associated with haploidentical HSCT. Recently, however, due to improvements in conditioning regimens and GVHD prophylaxis, reports have shown encouraging results, with TRM and OS rates similar to those of the previously preferred MRD and 10/10 MUD types [
11]. According to another report, MRDs and 10/10 MUDs were preferred since the OS was higher compared to alternative donor options (9/10 MUD and haploidentical donors) [
12,
13]. However, in our study, there was no significant difference in OS found between the 3 donor types (
P=0.102) or between the 10/10 HLA MUD and 9/10 HLA MUD groups (
P=0.870). The haploidentical donor group also showed promising results with 0 incidences of GVHD or VOD and 0 deaths up to the end point of our study. However, studies with a larger number of patients and longer follow-up on haploidentical donor recipients are needed.
In 2004, our institution reported a study that analyzed newly diagnosed AML patients from 1996 to 2003 and found 4 patients with
FLT3-ITD mutations. All 4 patients underwent the BH-AC-based induction regimen and achieved CR after induction. In all cases, however, relapse occurred and RFS and EFS were 0%. One patient relapsed after HSCT with an MRD, 2 relapsed before HSCT, and 1 was lost in follow-up [
6]. In this study, the OS, EFS, and RFS were 53.8±12.1%, 53.6±12.1%, and 72±12%, respectively, showing a significant improvement in outcomes for pediatric AML patients with
FLT3-ITD mutations over a period of 2 decades. This improvement in outcomes was also seen in other follow-up studies of AML patients with
FLT3-ITD mutations owing to advances in general medical care and aggressive treatment strategies, including earlier HSCTs [
3,
5,
8]. The CR rate after first-line induction therapy was 72.2% (13/18 patients) and the CR rate after salvage therapy was 80% (4/5 patients). The overall CR rate in this study was 94.4% (17/18 patients). Both studies from our institution showed a high overall response rate of 100% and 94.4%, respectively. A study in the adult population reported that 90% of patients achieved CR after 1 or 2 induction cycles [
14]. However, despite the high CR rate, more than half of the patients subsequently relapsed [
14]. In our previous study, the relapse rate was 100%. However, in this study, the relapse rate was 22%, which was lower than that reported in other studies. This could be due to our high rate of HSCT (94.4%). Taylor
et al. [
14] reported a high incidence of early relapse in AML patients with
FLT3-ITD mutations; however, when HSCT was performed at CR1, OS and remission duration significantly improved compared to when HSCT was not performed immediately. The primary reason for death in this study was relapse and disease progression (22%). All relapses in our study occurred after HSCT, with the time to relapse ranging from 4–28 months after HSCT. Our study showed relapses as late as 12 and 28 months after treatment, emphasizing the importance of off-therapy monitoring and suggesting the need for post-HSCT maintenance therapy in this population.
In this study, we detected the presence of
FLT3-ITD mutations but did not detect the mutation level, location, or size. Studies have addressed the association between allelic ratio and prognosis, reporting that patients with
FLT3-ITD mutations with a high allelic ratio had shorter OS, and patients with a longer inserted ITD length had a shorter RFS [
15,
16]. On the other hand, other studies have reported that
FLT3-ITD allelic ratio was not correlated with any survival outcome [
14,
17,
18]. Therefore, further studies are required to establish a more solid association between the
FLT3-ITD allelic ratio and survival rate. Cytogenetically normal AML has been regarded as a prognosis of intermediate risk, but the presence of
FLT3-ITD mutations within this group have been recognized to reduce RFS and OS compared to the FLT3-wt [
14]. However, the prognosis of
FLT3-ITD mutations in AML patients with abnormal karyotypes is unclear. In our study, 50% of patients had a normal karyotype by conventional cytogenetic analysis at diagnosis. Consistent with other studies, there were no significant differences in the OS or EFS for normal or abnormal karyotypes in patients with
FLT3-ITD mutations (
P=0.699) [
8,
19].
FLT3-ITD mutations coexisting with other specific molecular genetics and cytogenetic alterations have been frequently reported [
8]. Although not all patients were tested for these in this study, known concurrent cytogenetic abnormalities were identified in 7 patients (3 with
AML1/ETO rearrangement, 1 with
NPM1 mutation, 2 with
DEK/NUP214 fusion, and 1 with
MLL rearrangement). The
AML1/ETO rearrangement and
NPM1 mutation abnormalities are supposedly associated with a favorable prognosis [
20]. Consistent with previous reports, all of these 4 patients achieved CR1, none had relapses, and 3 continued to have no evidence of disease (NED) (1 died of TRM).
DEK/NUP214 fusions and
MLL rearrangements are usually associated with a worse prognosis [
20]; however, in our study, all 3 of these patients achieved remission, none relapsed, and 2 continued to have NED (1 died of TRM) (
Table 2). Consistent with our findings, the coexistence of
NPM1 mutations has been suggested to potentially ameliorate the poor prognosis of
FLT3-ITD in AML patients [
21]. However, the presence of
NPM1 mutation has not been associated with a significant difference in survival outcomes [
14,
21]. Data on the prognosis of combined
FLT3-TKD mutations remain unclear. In our study, patient 13 had a combined
FLT3-TKD mutation and had a good prognosis. The incorporation of routine concurrent analyses for cytogenetic abnormalities at diagnosis could provide a better understanding of potential prognostic implications in the future.
FLT3 inhibitors were not part of the treatment regimen in this study; however, these novel agents have been readily used and studied in the adult population and show promising results [
2]. In vitro studies have reported that FLT3 inhibitors work synergistically with chemotherapy to induce cytotoxicity [
22,
23]. Selective next generation FLT3 therapies (gliteritinib, crenolanib, quizartinib) have greater specificity for FLT3 and higher potency than multitargeted TKIs (midostaurin and sorafenib), and clinical trials have shown an improvement in OS even in relapsed/refractory patients [
2,
19]. This is promising because relapse is common in these patients, and relapsed patients usually present with higher
FLT3-ITD mutation burdens and have a poorer response to treatment [
24]. Sorafenib has been extensively studied as a potential post-HSCT maintenance therapy medication, with reports suggesting it was both well tolerated and effective in significantly reducing the incidence of relapse while improving survival after HSCT [
2,
25].
In summary, our study demonstrated a relapse rate of 22% after HSCT and OS and EFS rates of 53.8±12.1% and 53.6±12.1% in pediatric AML patients with FLT3-ITD mutations. This study was the first to focus on pediatric AML patients with FLT3-ITD mutations in Korea and outline their clinical course in a single institution over a decade. There were no significant differences in survival outcomes based on the induction chemotherapy regimens or the HSCT donor types in this study. Compared to our previous report from 2004, significant improvements in outcomes were seen, owing to advances in general medical care and aggressive treatment strategies, including earlier HSCTs. There are growing reports that FLT3 inhibitors are effective when added to induction, consolidation, salvage, and post-HSCT maintenance therapies. The incorporation of targeted FLT3 inhibitor therapies into current management strategies will hopefully improve the prognosis of this disease in the pediatric population. Thus, the significance of this study is that it can serve as a reference for future diagnostic (allelic ratio, molecular genetic testing) and therapeutic (FLT3 inhibitors) regimens yet to be incorporated into this high-risk pediatric population.
Go to :


 PDF
PDF Citation
Citation Print
Print


 XML Download
XML Download