

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Aplastic anemia (AA) is an unusual hematological disease defined by hypoplasia of the marrow and failure to form blood cells of all three lineages [1, 2]. The term AA is a misnomer as the disorder causes pancytopenia more than anemia [3]. The incidence of AA varies from 1.4 to 14 cases per million population [4]. The pathophysiology of AA was initially considered to be related to mere exposure to chemicals such as benzene or chloramphenicol. However, recent studies have shown that there are several other factors that cause immune dysregulation leading to AA [5]. The mechanism of immune dysregulation involves T-cell activation and cytokine production [6].
Recent studies have shown that defective functioning of regulatory T-cells leads to increased production of interferon gamma (IFN-γ) and tissue necrosis factor (TNF-α), causing stem cell injury, leading to bone marrow aplasia [7, 8]. Cytokine gene polymorphisms due to single nucleotide polymorphisms (SNPs) involved in AA are TNF-α and IFN-γ-874, the overproduction of which leads to immune destruction of hematopoietic stem cells (HSCs) [9]. An array of cytokines play a major role in the proliferation and differentiation of hematopoietic stem cells [10]. Two major cytokines, IFN-γ and TNF-α, are involved in the hematopoietic suppression and pathogenesis of acquired AA [11]. The human TNF gene is found on chromosome 6p21.3, spans approximately 3 kb, and contains four exons. It is an inflammatory cytokine. The TNF gene contains a single functional SNP, TNF-α-308 G/A, which is one of the most studied SNPs [6]. The human IFN-γ gene is found on chromosome 12q14, is a marker of immune system activity, and may itself be a mediator of bone marrow cell destruction. IFN-γ is a key pro-inflammatory cytokine that has a defensive role in host resistance against cancer and also contributes towards essential disease pathology. The IFN-γ gene contains two common polymorphisms, 874 A/T and 875 C/A, which may have functional products following gene transcription [12].
The current study was undertaken to evaluate cytokine gene polymorphisms in a North Indian population. We examined both cytokine gene polymorphisms (TNF-α-308 and IFN-γ-874) in our patients and in healthy controls to validate the genetic theory. In addition, we assessed the association of variants of these cytokines with clinical parameters, disease severity, and the response of patients with AA to immune suppressive therapy (IST).
MATERIALS AND METHODS
A case control study was carried out with AA patients attending the Clinical Hematology outpatient department (OPD) of King George’s Medical University (KGMU) in Lucknow, Uttar Pradesh, India, from March 2015 to August 2018.
Inclusion/exclusion criteria
Patients who were diagnosed with acquired AA were included in the study after obtaining their written informed consent. Subjects with bone marrow aplasia due to chemotherapy and/or radiotherapy, or bone marrow aplasia attributable to conditions such as PNH, Fanconi anemia, and hypoplastic MDS were excluded from the study. Subjects who were not willing to participate in the study were also excluded.
The diagnosis, classification (severe AA, non-severe AA, and very severe AA), and response assessment of AA were made following standard guidelines (Marsh et al., 2009; Camitta et al., 1975; Bacigalupo et al., 1988) [13].
Data collection
All data were collected using a predesigned questionnaire. Patients and/or their guardians were interviewed for data pertaining to the study, including demographic details and environmental factors. The study was approved by the Institutional Ethics Committee of KGMU.
Sample collection and genotyping
Five mL of peripheral blood was drawn into an ethylenediaminetetraacetic acid (EDTA) vial under aseptic conditions. Of this, 2 mL was used for genomic DNA extraction using Qiagen Kit (Qiagen, Hilden, Germany) and 3 mL was used for ELISA. The protocol followed that in the Qiagen instruction manual. Quality estimation of all extracted DNA samples was performed using 0.8% agarose gel electrophoresis. Genotyping of both SNPs (TNF-α-308 and IFN-γ-874) was performed using the PCR-restriction length fragment polymorphism method [14].
The primers used for the amplification of TNF-α-308 were: forward 5'-AGGCAATAGGTTTTGAGGGCCAT-3'; reverse, 5'-TCCTCCCTGCTCCGATTCCG-3', and primers used for the amplification of IFN-γ-874 were: forward, 5'-GATTTTA TTCTTACAACACAAAATCAAGAC-3'; reverse, 5'-GCAAA GCCACCCCACTATAA-3'.
The PCR reaction was performed using a DNA thermal cycler (Eppendorf Mastercycler Nexus Thermal Cyclers, Hamburg, Germany). PCR amplification was carried out on a final sample volume of 20 mL (3 mL DNA, 10 mL Top Taq PCR Master Mix, 1 mL primer; each forward and reverse, and 5 mL distilled water). The thermal cycler was programmed as follows for the different genes: TNF-α-308 gene polymorphism, PCR conditions were the same except for a change in annealing temperature to 62.3°C for 45 seconds; IFN-γ-874 gene polymorphism, the annealing temperature was 58°C for 45 seconds. The amplified PCR product was then electrophoresed on a 2.0% agarose gel.
Digestion of the amplified product by specific restriction enzymes for each polymorphism
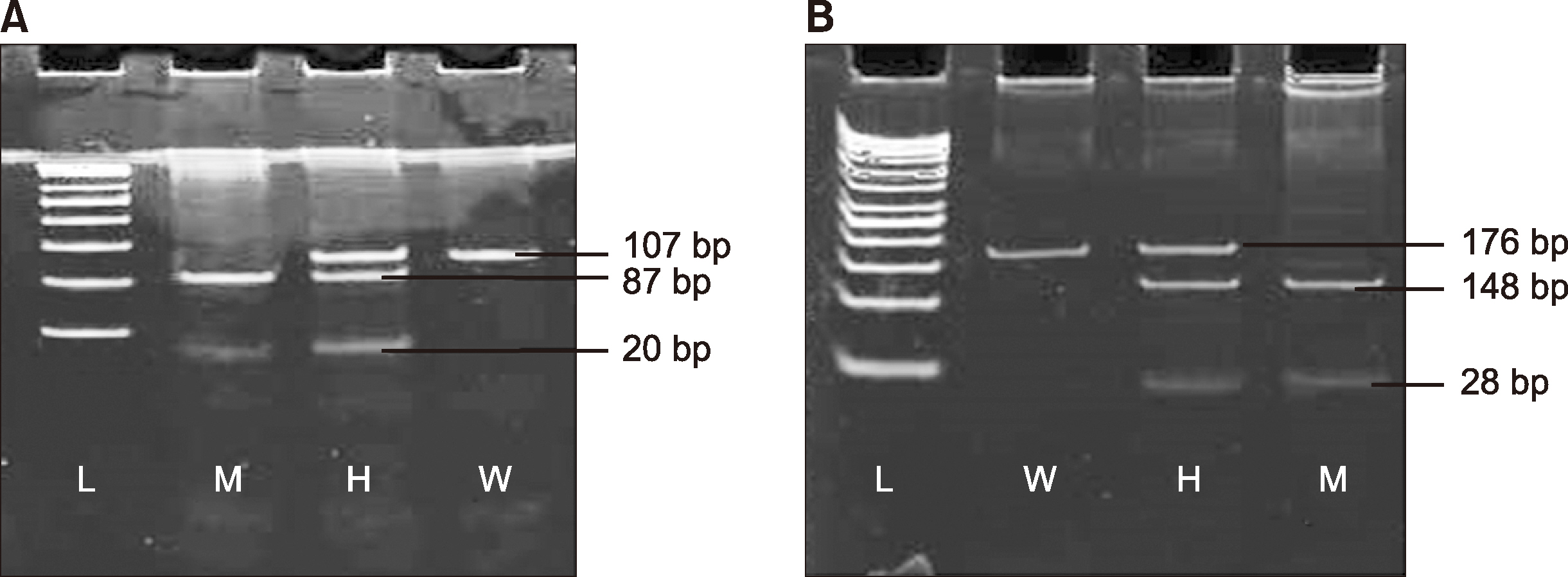
The amplified PCR product (10 mL) mixed with a restriction enzyme (1 mL; New England Biolabs, Hitchin, UK) was used in the reaction. The reaction mixture was incubated for 2 h at 37°C. The digested products underwent gel electrophoresis in the range of 1.5–3%. The separated fragments were then stained with EtBr and visualized along with a ladder using the molecular imager gel doc XR System (Bio-Rad, Hercules, CA, USA). The details of the restriction enzymes and their resulting base pair lengths are shown in Table 1. The gel pictures of TNF-α-308, IFN-γ-874 gene polymorphisms are shown in Fig. 1.
Measurement of plasma TNF-α and IFN-γ levels
Three mL of peripheral blood was drawn into an EDTA-containing vial. For isolation of the plasma, samples were centrifuged for 15 min at 2,500×g. Plasma levels of TNF-α and IFN-γ were evaluated using a commercially available ELISA kit (Abcam, Cambridge, MA, USA). This kit was used to identify cytokines using specific monoclonal antibodies according to the manufacturer’s instructions.
Statistical analysis
All data were double-entered in Microsoft Excel. Statistical analysis was performed using SPSS (version 17.0, SPSS Inc., Chicago, IL, USA). Univariate analysis was performed for categorical variables expressed as percentage and frequencies, and the mean and standard deviation was calculated for continuous variables. The chi-square test was used for categorical variables and the Hardy-Weinberg Equilibrium (HWE) was analyzed for all cases and controls separately. Bivariate logistic regression analysis was used to calculate the odds ratio (OR). Statistical significance was defined as P<0.05. Three genetic models were used to determine the significant association between the genotypes and AA. In the dominant model, the wild-type homozygous genotype was compared with the union of heterozygous and mutant genotypes. In the over-dominant model, the combined values of wild-type homozygous and mutant homozygous were compared with those of the heterozygous genotype. In the recessive model, mutant homozygous was compared with the combined values of heterozygous and wild type homozygous.
RESULTS
Two hundred and forty subjects were enrolled in the study (120 patients 120 and healthy controls). The ratio of male to female AA patients was 70:30. Severity was categorized as: non-severe AA (NSAA), 46.7%; severe AA (SAA), 44.2%; and very severe AA (VSAA), 9.1%. The demographic variables and clinical features of AA patients and healthy controls are summarized in Table 2.
Evaluation of TNF-α-308 and IFN-γ-874 polymorphisms and susceptibility to developing AA
Individuals with the IFN-γ-874 genotype showed significant susceptibility to developing AA, while the TNF-α-308 genotype was found to be protective, with carriers demonstrating a lower risk of developing AA (Table 3). Carriers of the heterozygous (GA) TNF-α-308 genotype showed a lower risk, and carriers of the heterozygous (AT) IFN-γ-874 genotype showed a significantly higher risk of developing AA relative to healthy controls [OR, 0.56; 95% CI, 0.32–0.98; P=0.043; (OR, 1.76; 95% CI, 1.00–3.10; P=0.047, repsecitvley)]. Table 3 depicts the genetic models for a polymorphism (IFN-γ-874) for which a significant association was observed in the over-dominant model (AT vs. AA+TT) (OR, 1.80; 95% CI, 1.05–3.09; P=0.030).
Evaluation of TNF-α-308 and IFN-γ-874 polymorphisms in NSAA, SAA, and VSAA
Notably, no significant association was observed between the TNF-α-308 genotype and severity of AA (Table 4). The frequency of the heterozygous (AT) IFN-γ-874 genotype demonstrated significant association with NSAA vs. control (OR, 2.06; 95% CI, 1.00–4.22; P=0.046). On the other hand, Table 4 shows the possible genetic models among the groups with different AA severity, which showed no significant association with the severity of AA or controls.
Evaluation of TNF-α-308 and IFN-γ-874 polymorphisms in response to IST
No significant association between TNF-α-308 or IFN-γ-874 and response to IST in patients with acquired AA was observed (Table 5). Similarly, in genetic models, a comparison was made between complete and partial responders with non-responders, and none of which were found to be significant (Table 5).
Plasma concentrations of TNF-α-308 and IFN-γ-874 in patients with AA and controls
TNF-α and IFN-γ plasma levels were higher in 70.8% and 77.5% of patients with AA relative to those in controls, respectively. The highest levels were observed in VSAA patients, with a mean of 76.35±1.95 pg/mL for TNF-α and 81.23±2.01 pg/mL for IFN-γ. Significantly higher TNF-α and IFN-γ concentrations were also observed in SAA patients, with a mean of 25.71±1.81 pg/mL for TNF-α and 28.29±1.89 pg/mL for IFN-γ. No significant difference was observed in NSAA patients (Fig. 2).
DISCUSSION
The pathogenesis of AA involves changes in cell susceptibility, damage to HSCs, and development of an abnormal hematopoietic environment, which might result in bone marrow failure [7]. Dysfunction of cytokines and T-cell subsets might be key reasons for the development of AA [15-17]. The mechanisms involved in AA primarily comprise a cell-mediated killing and discharge of T-helper cytokines, such as IFN-γ and TNF-α, with an inhibitory effect on NK cells [18, 19]. To evaluate the association of cytokine gene polymorphisms with susceptibility, severity, and response to IST, we studied the IFN-γ-874 and TNF-α-308 polymorphisms and found that IFN-γ-874 may be involved in the development of AA. The IFN-γ-874 gene was found to be strongly associated with susceptibility to developing AA and disease severity, which is in agreement with earlier research on the subject [20, 21]. TNF-α-308 positively correlated with AA in our population; this result is not in agreement with earlier findings available on the subject [22]. This inconsistency might be due to the smaller sample size, selection criteria, ethnicity, and geographical variation of study subjects.
Several studies on the role of polymorphisms in AA have been carried out, but this is the first study from North India on the subject. As predicted, the IFN-γ-874 gene polymorphism showed similar results as reported in the literature [21]. No association was recorded in both the inflammatory cytokines after IST between AA and with those of control. Our results are in disagreement with previous reports that showed an association after IST [23, 24].
A significant number of AA patients had increased levels of TNF-α and IFN-γ in their blood plasma, and these levels were significantly higher in patients in the severe and very severe groups than in the control group. This is the first study performed in India showing elevated TNF-α and IFN-γ levels in the blood of AA patients.
AA is a rare blood disorder, and this study is important as it is the first study from north India. Limitations of the study include the small sample size, selection criteria, ethnicity, and geographical variation of the population involved in the study.
In conclusion, our results suggest that polymorphism in the IFN-γ-874 gene is associated with the development of AA, but there is no such association for TNF-α-308. Furthermore, these results conclusively indicate that polymorphisms affecting cytokine gene polymorphisms will be helpful in the understanding of AA. Additional studies are required with a larger sample size to validate that cytokine gene polymorphism and its strong association with AA and response to IST.

 XML Download
XML Download