

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Particulate air pollution is an environmental health risk factor that is associated with increased cardiovascular morbidity and mortality. Epidemiologic studies have recommended that elevated concentrations of ambient particulate matter (PM) are strongly associated with increases in mortality, hospital admissions, episodes of acute ischemia, and arrhythmia.1)2)3)4)
Common components of PM contain nitrates, sulfates, elemental and organic carbon, organic compounds (e.g., polycyclic aromatic hydrocarbons), biological compounds (e.g., endotoxins, cell fragments), and various metals (e.g, iron, copper, nickel, zinc, and vanadium).5) Exposure to PM produces systemic inflammation, endothelial damage and autonomic alteration that develops into cardiovascular diseases.6) The risk of mortality associated with life-threatening arrhythmias increases in relation to PM10 (particles measuring 10 μm or less).7)8)9) In most studies, fine particular matter <2.5 μm in aerodynamic diameter (PM2.5) is consistently associated with sudden cardiac death, heart failure, and myocardial infarction.1)3)4) Despite evidence for arrhythmogenic action of PM, the underlying arrhythmogenic mechanisms of PM are still poorly understood.
Oxidative stress increases after exposure to raised levels of ambient particles.10) Ca2+/calmodulin-dependent protein kinase II (CaMKII) is activated by intensified intracellular Ca2+ from β-adrenergic receptor stimulation.11) However, it was recently disclosed that oxidation of paired regulatory domain methionine residues sustains CaMKII activity in the deficiency of Ca2+/CaM.12) CaMKII activation also prolongs action potential duration (APD) and induces afterdepolarization in cardiomyocytes, by impairing I
Na inactivation and enhancing I
CaL. In a previous study, we found that intracoronary circulation of diesel exhaust particles could cause arrhythmia via oxidative stress and CaMKII activation. Although several studies showed the relationship between ambient particular matter and arrhythmia, the effect of inhaled ambient PM on arrhythmia has not been well elucidated.13)14) Moreover, the mechanism by which inhalation of PM induces arrhythmias and cardiovascular disease has not been evaluated. This study investigated the threshold level to induce arrhythmia, as well as arrhythmogenic mechanism of real urban air pollution (RUA) that mostly consisted of PM2.5 close to ultrafine particles (UFPs).
METHODS
This study's protocol was supported by the Institutional Animal Care and Use Committee of Yonsei University (IACUC-201710-640-01), and conformed to the guideline for the care and use of laboratory animals published by the United States National Institutes of Health.
Particle characterization
Soot particles were produced using a newly developed pyrolysis-based RUA generator.15) Particle morphology, chemical composition, and size distribution were known to be very similar to the PMs emitted from automotive engines and industrial combustors. In this experiment, propylene (C3H6) gas was dispersed in nitrogen, and its mole fraction was 0.014. Gas mixture was heated to ~1,300oC within an alumina tube surrounded by electrical heaters. The fuel then underwent a soot formation process, which generally includes nucleation, surface growth, and agglomeration steps. Exhaust gas from the device was free of NOx and CO, since the fuel was carbonized under an oxygen-free atmosphere (Figure 1A, Supplementary Figure 1). Individual spherules were measured by ImageJ software. Transmission electron microscope (TEM) (JEOL, JEM-2100F, Tokyo, Japan) image of the soot particles is shown in Figure 1B. Aerodynamic size of the aggregates, which determines PM class, was measured by a particle size analyzer (TSI, Nanoscan SMPS 3910, Saint Paul, MN, USA). The mean sizes±standard deviation were 205 nm and 78 nm. Particles were composed of PM2.5 that were close to UFPs (Figure 1C).
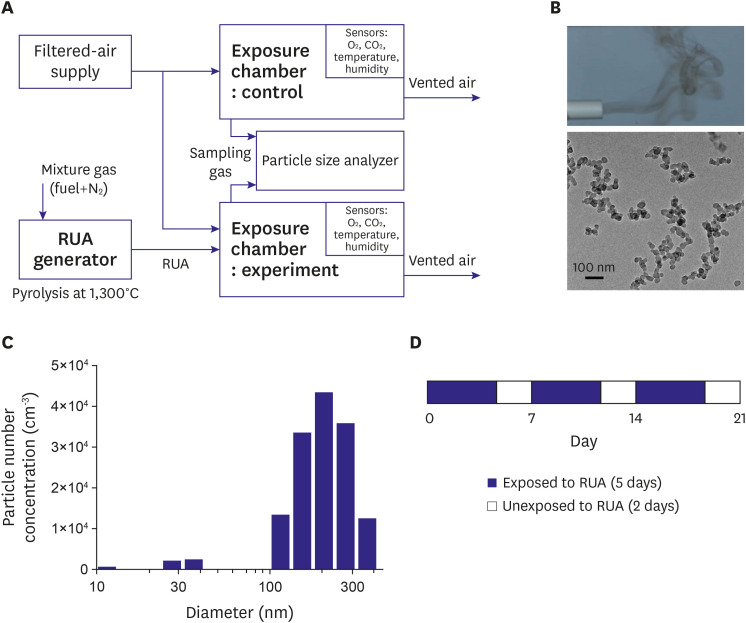
Exposure chamber and animal model
Two identical acrylic chambers (1 m×1 m×0.8 m) were manufactured to provide whole-body exposure (Supplementary Figure 1). Chambers were air-tight with the doors closed. Filtered air was supplied to gas inlet ports installed on the tops of chambers, in order to maintain O2 level and to limit maximum CO2 concentration in chambers. A portion of the exhaust gas from soot generator was taken and delivered to the control group chamber alone through another inlet port on the top. The air inside chambers was continuously circulated and homogenized using fans. Outlet ports on the side walls were left open so that the ventilating air continued to flow outward due to the slight positive pressure built up inside chambers. Temperature, O2 concentration, CO2 concentration, and humidity were continuously monitored with sensors. RUA concentration was measured every 20–30 min using particle size analyzer, and data were cross-checked with measurement results from a multi-wavelength Aethalometer (AE-42, Magee Scientific, Berkeley, CA, USA). Table 1 summarizes the key parameters of exposure process. Experiments for control and RUA of 800 μg/m3 were performed under the same condition. Experiments for RUA of 200 μg/m3 and RUA of 400 μg/m3 were also performed under the same condition.
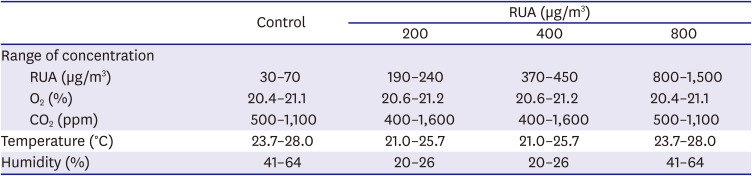
Table 1
The conditions of the atmosphere inside the exposure chambers
*The experiments for control and RUA of 800 μg/m3 were performed at a same condition. And the experiments for RUA of 200 μg/m3 and RUA of 400 μg/m3 were performed at a same condition.
RUA = real urban air pollution.
![]()
We obtained 6–8-week-old male C57BL/6 mice. C57BL/6 male mice were divided into four groups: a control group (control, n=12) and three groups with exposure to RUA with the concentration of 200 µg/m3 (n=12), 400 µg/m3 (n=12), and 800 µg/m3 (n=12). Mice were exposed to RUA at each concentration for 8 hr/day and 5 day/week to mimic human activity (Figure 1D). The overall duration of exposure was 3 weeks.
Optical mapping and experimental protocol
Mice were anesthetized with intraperitoneal injection of ketamine (80 mg/kg) and xylazine (4 mg/kg). An electrocardiogram (ECG) was consecutively noted for 30 min. The ECGs were manually evaluated by a cardiologist. QT intervals were executed from Q to the end of T for 10 beats, and then averaged. ECG was measured in 12 mice in each group.
Chests of mice were opened via median sternotomy, and their hearts were quickly excised and immersed in cold Tyrode's solution (composition in mmol/L: 125 NaCl, 4.5 KCl, 0.25 MgCl2, 24 NaHCO3, 1.8 NaH2PO4, 1.8 CaCl2, and 5.5 Glucose). The ascending aorta was cannulated and perfused with 37°C Tyrode's solution equilibrated with 95% O2 and 5% CO2 to maintain a pH of 7.4. The heart motion was inhibited by 10–17 µmol/L of blebbistatin. Then, hearts were stained with Rhod-2 and RH237 (Molecular Probes, Eugene, OR, USA), and excited with laser light at 532 nm.16) Fluorescence was recorded using two cameras (MiCAM Ultima, BrainVision, Tokyo, Japan) at 1 ms/frame and 100×100 pixels, with spatial resolution of 0.35×0.35 mm2/pixel. APD was measured from (dF/dt) max to 90% recovery to baseline, APD. Calcium transients (CaD) was chosen from the maximum first derivative of Cai upstroke to the time point of 90% recovery of Cai to its normal baseline. Local conduction velocity (CV) vectors were computed for each pixel from the differences in activation time-points for that pixel (determined from [dF/dt] max) and its 7×7 nearest neighbors, as previously described.16)17)
To test ventricular tachycardia (VT) or ventricular fibrillation (VF) vulnerability, the heart was paced at the left ventricle (LV) using a programmed stimulation protocol consisting of 20 S1 pulses at 250 ms cycle length (CL), followed by a premature S2 pulse with continuously shorter S1-S2 interval steps: 250 to 100 ms in 20 ms steps, 100 to 70 ms in 10 ms steps, and 60 to 35 in 5 ms steps, until loss of capture or initiation of VT. VT/VF inducibility was defined as the ability to provoke VT/VF with the pacing protocol. Sustain ventricular arrhythmia was defined as duration longer than 30 seconds. Electrophysiology studies were performed in 10 mice in four group.
Histology and immunoblot analysis
The hearts were in 10% formalin, embedded in paraffin, and stained Masson's trichrome for histologic estimation. Quantification of fibrosis areas was showed as the percentage of stained area compared to the total area of fields examined, using ImageJ software (National Institutes of Health, Bethesda, MD, USA).
To evaluate the level of fibrosis, inflammation and oxidative stress in heart tissues, immunoblotting of antigens were examined with the following primary antibodies: anti-transforming growth factor-beta (TGF-β) (1:1,000, Abcam Reagents, Cambridge, MA, USA), anti-MMP2 (1:1,000, Abcam Reagents), anti-MMP9 (1:1,000, Abcam Reagents), anti- collagen-I (ColI) (1:1,000, Abcam Reagents), anti-collagen-III (ColIII) (1:1,000, Abcam Reagents), anti-interleukin-6 (1:1,000, Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-Cox-2 (1:1,000, Santa Cruz Biotechnology), anti-TNF-a (1:1,000, Abcam Reagents), anti-iNOS (1:1,000, Santa Cruz Biotechnology), and HMGB1 (1:1,000, Abcam Reagents). The protein levels of total Ca2+/calmodulin-dependent protein kinase II δ (CaMKIIδ) and CaMKII-Thr287 (1:1,000; Abcam Reagents), GAPDH (1:100,000; Abcam Reagents), total phospholamban (PLB) (1:1,000; Abcam Reagents), Thr17 phosphorylated PLB (1:1,000; Santa Cruz Biotechnology), calsequestrin 2 (CSQ2) (1:1,000, Santa Cruz Biotechnology), and NCX (1:1,000, Santa Cruz Biotechnology) were quantified by western blot. Bands were scanned, and their intensities were quantified using Image J software.
Assay of inflammatory cytokines
Blood was obtained from the abdominal aorta in the four groups on day 21. Enzyme-linked immunosorbent assay was executed to establish the expression levels of TNF-α, IL-6, and HMGB1 in serum. By the manufacturer's instructions, protein levels in serum were quantified with TNF-α (R&D System, Minneapolis, MN, USA), IL-6 (R&D System), and HMGB1 (IBL International, Hamburg, Germany) kits.
Immunohistochemical staining
For immunostaining of heart tissue, anterior walls of left ventricles were harvested after being fixed with 10% formaldehyde in PBS (pH 7.4), combined with paraffin, and then sectioned at a thickness of 4 µm. immunohistochemistry (IHC) was discovered with avidin-biotin complex (ABC) method. The primary antibodies used were mouse anti-CD68 monoclonal antibody (1:2,000, Abcam Reagents), finding only CD68. Inactive tissue endogenous peroxidase was then blocked by incubation with peroxidase block for 30 minutes and sections were incubated for enzymatic retrieval, followed by 10% blocking serum. Immunostaining was colored using polymer method (Vectastain ABC kits, PK-4000, Vector Laboratories, Burlingame, CA, USA), and later color development was completed with diaminobenzidine.
Statistical analysis
Data were expressed as the mean±SEM. ANOVA tests with post-HOC and Bonferroni's correction were used to compare the means among four groups. Pearson's chi-squared tests were used to compare 2 categorical variables. Analysis was performed using the statistical software package, SPSS version 23.0 (IBM, Armonk, NY, USA). P-value<0.05 was considered statistically significant.
RESULTS
EKG changes after exposure to RUA
Figure 2A shows typical EKGs of control and RUA groups. The QRS and QTc intervals were prolonged gradually with the increase of RUA concentration. Compared to control mice, RUA of 400 µg/m3 and 800 µg/m3 significantly decreased heart rate, and prolonged QRS and QT intervals (all p<0.05) (Figure 2B). Figure 2C shows premature ventricular contractions (PVCs) (upper panel), as well as non-sustained (middle panel) and sustained ventricular tachycardia (VT) (lower panel) documented in RUA-exposed mice. While no PVC was observed in control, PVCs were observed in 17% of mice exposed to RUA of 400 µg/m3. Significant increases of spontaneous VT or VF were observed in 42% of mice exposed to RUA of 800 µg/m3 (p<0.001) (Figure 2D).
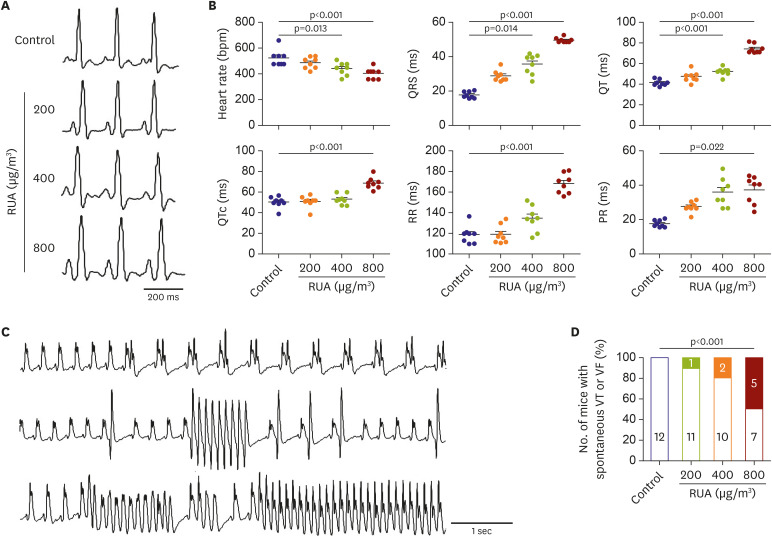
Figure 2
RUA dose-dependent increase of QT intervals and ventricular arrhythmia. (A) Typical examples of electrocardiogram lead I. (B) Heart rate, QRS, QT, QTc, RR and PR intervals. Data are expressed as mean±standard error of the mean. (C) Spontaneous premature ventricular contractions (upper panel), non-sustained (middle panel), and sustained ventricular tachycardia (lower panel) during RUA exposure. (D) Dose-dependent increase of mouse with spontaneous VT or VF in RUA group (n=12 per group).
RUA = real urban air pollution; VF = ventricular fibrillation; VT = ventricular tachycardia.
![]()
APD prolongation and increased apicobasal repolarization gradient in RUA-exposed mice
Typical traces of Vm (black) and Cai (red) recorded from LV of control and RUA mouse heart at the pacing CLs of 200 ms (Figure 3A). APD and CaD were prolonged gradually with the increase of RUA concentration. Compared to control, APD and CaD were significantly increased from RUA concentration of 400 µg/m3 at the pacing CLs of 200 ms (p<0.001) (Figure 3B).
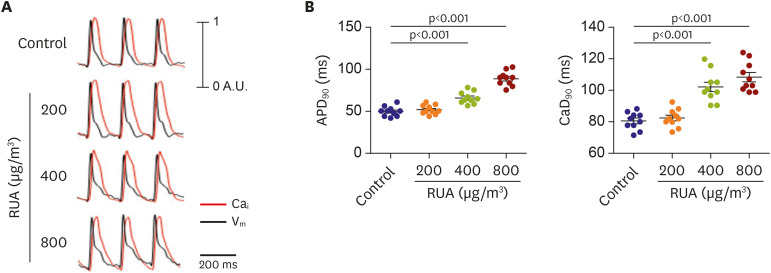
Figure 3
RUA dose-dependent increase of APD and CaD in RUA group. (A) Typical action potential traces of voltage (black) and calcium (red) at the pacing cycle length of 200 ms. (B) Comparison of APD and CaD among various groups. Note a RUA dose-dependent increase of APD and CaD (n=10 for each group tested). Data are expressed as mean±standard error of the mean.
APD = action potential duration; CaD = calcium duration; RUA = real urban air pollution.
![]()
Figure 4A shows the activation (left panels), APD (middle panels), and CV (right panels) maps from control and RUA of 800 µg/m3-exposed mouse hearts. Compared to controls, RUA-exposed mouse hearts showed slower activation, and higher increase in APD and CV heterogeneity. Figure 4B shows Vm tracing recorded at the base and apex of mouse heart. The apicobasal repolarization gradient, which was measured as the difference in APD between base and apex, was dose-dependently increased in RUA-exposed mice, and significantly increased from RUA concentration of 400 µg/m3 than control at the CL of 300, 200, and 160 ms (all p<0.05) (Figure 4C). Apicobasal reentry was observed in RUA-exposed mouse hearts (Supplementary Figure 2). Finally, VT or VF was easily induced at a longer pacing CL with the increase of RUA concentration, and significantly increased from RUA concentration of 400 µg/m3 compared to control (p=0.009) (Figure 4D).
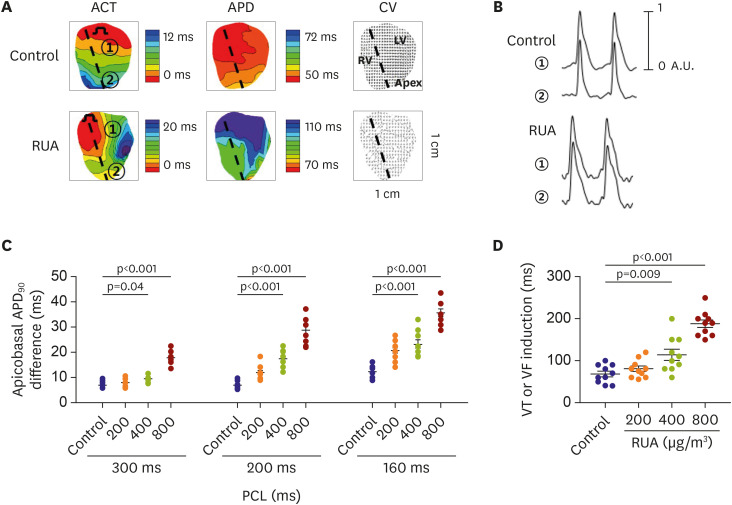
Figure 4
RUA dose-dependent increase of apicobasal APD90 difference and ventricular arrhythmia inducibility. (A) Activation, action potential duration, and conduction vector maps in control and in mouse exposed to RUA of 800 μg/m3. Dotted line marks interventricular septum. Base (①) and apex (②) of left ventricle are action potential recording sites. (B) Action potential tracings recorded from base (①) and apex (②) of left ventricle at the pacing cycle length of 200 ms in Langendorff-perfused mouse heart. (C) RUA dose-dependent increase of apicobasal APD difference. (D) RUA dose-dependent increase of VT or VF inducibility (n=10 for each group tested). Data are expressed as mean±standard error of the mean.
ACT = activation; APD = action potential duration; CV = conduction velocity; LV = left ventricle; RUA = real urban air pollution; RV = right ventricle; VF = ventricular fibrillation; VT = ventricular tachycardia.
![]()
Increased inflammatory cell infiltration and fibrosis in RUA-exposed mice
IHC staining showed CD68 immunoreactivity in LV of mouse heart tissues of RUA-exposed compared to control mice. Interestingly, the greatest increase in immune cells was observed gradually with the monocyte population in RUA concentration of 400 µg/m3 (5.8±1.1 vs. 1.6±0.2%, p<0.001) (Figure 5A).
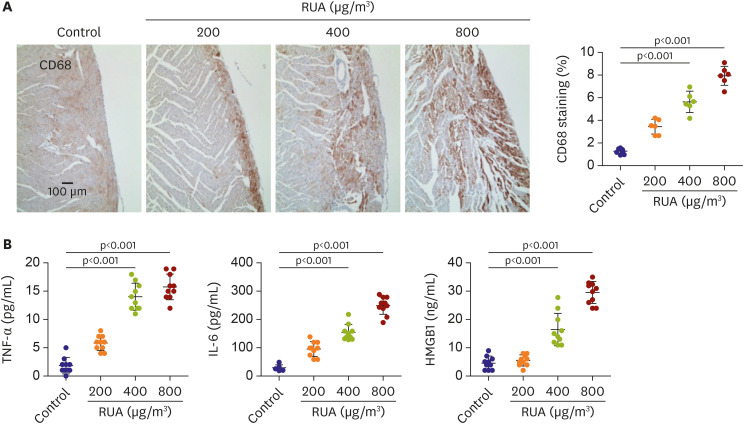
Figure 5
RUA dose-dependent increase of inflammation. (A) Typical CD68 DAB stain images of hearts at each dosage of RUA. (B) TNF-α, IL-6 and HMGB1 in serum (n=10 for each group tested). Data are expressed as mean±standard error of the mean.
HMGB1 = high-mobility group protein B1; IL = interleukin; RUA = real urban air pollution; TNF = tumor necrosis factor.
![]()
On ELISA, RUA group had increased serum levels of TNF-α (14.1±2.0 vs. 2.8±1.9, p<0.001), IL-6 (160.1±29.4 vs. 20.5±11.9, p<0.001), and HMGB1 (16.1±5.0 vs. 5.1±2.3, p<0.001) than controls (Figure 5B). Therefore, it can be concluded that these inflammatory markers increase progressively in RUA group.
Mice subjected to RUA for 3 weeks developed profound ventricular remodeling associated with cardiac fibrosis. The results showed that the fibrotic areas of cardiac and perivascular fibrosis were increased in RUA-exposed mouse compared to control mice (Figures 6A and B). At the same time, to further evaluate molecular change of TGF-β, MMP2, MMP9, fibrous structural proteins, collagen I and III were detected in heart tissues (Figure 6C). These factors were closely associated with myocardial fibrosis and detected by western blot. Data showed that the protein expression levels of TGF-β, MMP2, MMP9, collagens I and III were significantly increased in RUA-exposed group compared to control group (Figure 6D).
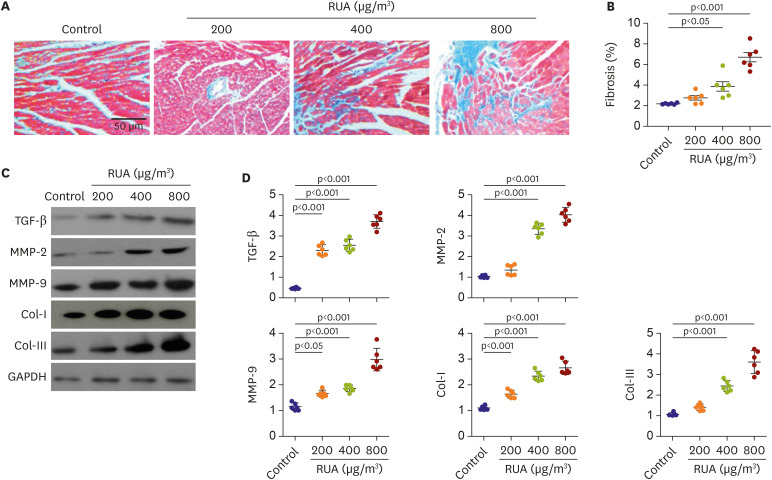
Figure 6
RUA dose-dependent increase of fibrosis. (A) Images of Masson trichrome staining patterns of heart at each dosage of RUA. (B) Scatterplots show quantification of the percentage of fibrotic areas in the histological sections. (C, D) Protein expression levels of TGF-β, MMP-2, MMP-9, Col-Iand Col-III were detected by western blotting and were quantified using GAPDH as an internal reference.
RUA = real urban air pollution; TGF-β = transforming growth factor-beta.
![]()
Moreover, Cx43 expression was also significantly decreased in RUA-exposed mice compared to control mice (1.0±0.5 vs. 0.5±0.1, p<0.001) (Supplementary Figure 3).
RUA increases oxidative stress and CaMKII activation
Compared to controls, IL-6, Cox-2, TNF-α, iNOS, and HMGB1 expression levels were increased by 3.2 (p<0.001), 2.5 (p=0.003), 4.2 (p=0.005), 1.4 (p<0.001), and 1.9 (p<0.001) times in LV from mice that were exposed to RUA concentration of 800 µg/m3, respectively (Figure 7A).
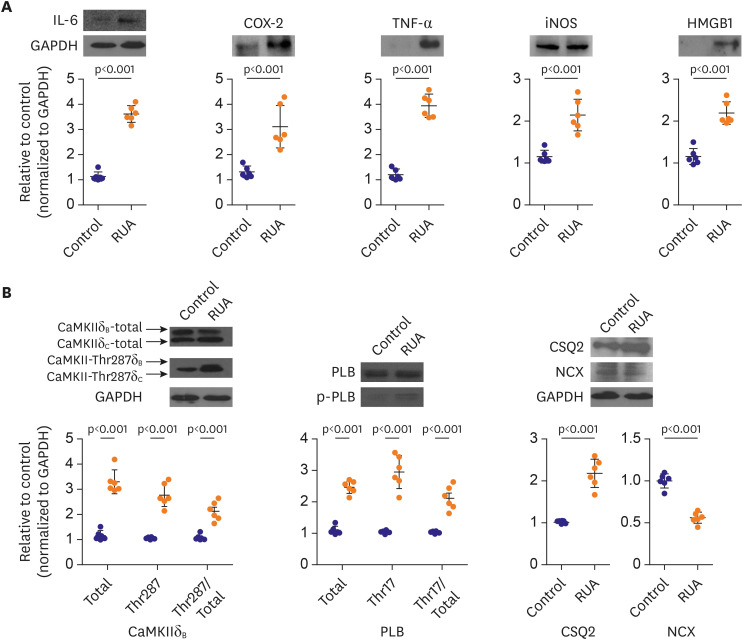
Figure 7
Exposure to RUA of 800 μg/m3 increases inflammation, oxidative stress, and phosphorylation of Ca2+-handling proteins. (A) Western blot-based expression analysis of IL-6, COX-2, TNF-α, iNOS, and HMGB1 proteins (upper panels), and quantification in left ventricular tissues from each group (lower panels). (B) Level of autophosphorylated CaMKII at Thr287, PLB, CSQ2, and NCX in left ventricular tissue from each group. Data are expressed as mean±standard error of the mean.
CaMKII = calmodulin-dependent protein kinase II; CSQ2 = calsequestrin 2; HMGB1 = high-mobility group protein B1; IL = interleukin; PLB = phospholamban; RUA = real urban air pollution; TNF = tumor necrosis factor.
![]()
We estimated CaMKII, PLB, CSQ2, and NCX expression levels in mouse ventricular tissue with western blotting. CaMKII at Thr287 (224%), phosphorylated PLB at Thr17 (263%), and CSQ2 (180%) were increased in LV from mice that were exposed to RUA concentration of 800 µg/m3, compared to control (p<0.001). NCX was decreased in RUA-exposed mouse hearts compared to those in control group (p<0.001, Figure 7B).
DISCUSSION
The main finding of this study is that EKG changes, including QRS and QTc intervals, were aggravated with the increase of RUA concentration. EKG change and arrhythmia increased significantly from the exposure to RUA concentration of 400 µg/m3. RUA of 400 µg/m3 increased apicobasal repolarization gradient and inducibility of ventricular arrhythmia, with increase of fibrosis. Finally, we have demonstrated that RUA-exposed mice had increased inflammation, oxidative stress, and CaMKII activation.
Threshold of EKG change and ventricular arrhythmia due to RUA
Epidemiological studies corroborate the elevated risk for cardiovascular events associated with exposure to PM2.5.18)19) PM2.5 generally has been associated with increased risks of myocardial infarction, stroke, arrhythmia, and heart failure exacerbation within hours to days of exposure in susceptible individuals. Some new studies have also proved that living in locations with higher long-term average PM levels elevates the risk for cardiac events. Recent evidence have also implicated other size fractions, such as UFPs <0.1 µm, gaseous copollutants (e.g., ozone and nitrogen oxides [NOx]), and specific sources of pollution (e.g., traffic).5) The U.S. Environmental Protection Agency (EPA) strengthened the National Ambient Air Quality Standards (NAAQS) for daily PM2.5 levels starting in 2006 (down from 65 to 35 µg/m3). Most recent NAAQS for Criteria Air Pollutants for PM2.5 is the annual mean of 15 µg/m3 and 24 hour of 35 µg/m3.
In this study, the changes of EKG, arrhythmias, and fibrosis of LV were observed from RUA concentration of 400 µg/m3 with exposure protocol of 8 hr/day and 5 day/week. This dosage is consistent with 24-hour exposure to 95 µg/m3, which is about three times higher than daily PM2.5 levels of U.S. EPA. Oxidative stress and CaMKII activation were observed in RUA concentration of 800 µg/m3 with exposure protocol of 8 hr/day and 5 day/week, which is consistent with 24-hour exposure to 190 µg/m3, and about six times higher than daily PM2.5 levels of U.S. EPA.
Mechanism of ventricular arrhythmia due to RUA
In this study, RUA-exposed mice showed electrophysiological features characterized by increased QTc intervals and APD, as well as spontaneous ventricular arrhythmia. In Langendorff-perfused mouse hearts, RUA-induced APD prolongation was mostly studied at the base of LV in this study, sparing the apex. This resulted in important apicobasal repolarization gradients, a finding that was consistent with previous studies.13)16) Also, improved dispersion of repolarization is crucial for inducing arrhythmia.20) Transmural and apicobasal dispersion of repolarization was shown to be responsible for the initiation of reentrant activation in patients. It has been reported that different mammalian species, including humans, have apico-basal differences in cardiac repolarization. The current study proposes that RUA exposure can prolong QT intervals and induce arrhythmia in susceptible patients. Lately, Sivagangabalan et al.21) showed that concentrated PM and O3 can alter dispersion of ventricular repolarization in healthy volunteers. We believe that our study might help describe the mechanism for dispersion of repolarization after exposure to PM. The RUA-exposed mice also showed CV heterogeneity. Increased level of fibrosis and reduction of Cx43 might be responsible for conduction disturbance.
Increased oxidative stress and phosphorylation of Ca2+-handling proteins in RUA-exposed mice
In this study, hearts from RUA-exposed mice showed increased inflammation and oxidative stress. Despite on-going debate regarding which particle components are responsible for producing ROS, there is collecting proof that pro-oxidative organic hydrocarbons, such as polycyclic aromatic hydrocarbons and quinones, and transition metals play a major role.22) Increased ROS generation is thought to induce excessive oxidative stress and impair endothelial-dependent vascular homeostasis. A number of previous studies have assumed that inhaled PMs caused oxidative stress and production of ROS, which are involved in the pathogenesis of cardiovascular diseases including hypertension, atherosclerosis, and endothelial dysfunction.23)24)
Even though our effect estimates were generally inconsistent in atherosclerosis or the systemic inflammation, we recorded higher estimates in highly exposed RUA group. The low precision of our estimates prohibit definite conclusion about factors. However, our findings basis further investigations of specific mechanisms and other characteristic.
Added to the enhanced activation due to increased intracellular Ca2+ levels from β-adrenergic receptor stimulation, it has been revealed that CaMKII activity is also enhanced by pro-oxidant conditions.12)25) Interestingly, the L-type Ca2+ channel is a major regulator of Ca2+ homeostasis and has been implicated in the genesis of arrhythmia. In addition to, Oxidation of paired regulatory domain methionine residues sustains CaMKII activity in the absence of Ca2+/CaM.12) These results support another study, which reported that ROS induces APD, Ca2+ duration prolongation and advances afterdepolarization in cardiomyocytes.26) We could not present phosphorylated PLB at ser16, our in vivo experiments consistently suggest that the mechanism of arrhythmia are mediated by oxidative stress and CaMKII activation.
Study limitations
This study has some limitations. It was proposed that inhaled nano-sized particles in air pollution can transmigrate across the human pulmonary epithelium into the systemic arterial circulation.27)28) This study provided more light on the associations between inflammation and fibrosis in cardiac tissues. Numerous experimental study in animals has already shown negative associations between air pollution and serum level.29) Thus, we measured ECG without sacrifice between animal groups, but not serum level. Additionally, more studies are required to further define the role of systemic inflammation and atherosclerosis in RUA exposed model. However, the actual concentration of particles in blood after inhalation could not be evaluated in this study. Second, RUA was higher in our study than in physiological condition. Although our study cannot describe the relationship between air pollution and arrhythmia under normal conditions, this study consistently suggests that RUA concentration of more than three times the daily PM2.5 levels of U.S. EPA can induce cardiac change and arrhythmia. Third, the ECG of the mouse was difficult to confirm, but the ventricular arrhythmia was well distinguished, but the atrial arrhythmia was indistinguishable. Previous studies have shown that esophageal lead induces atrial arrhythmia. Additionally, bradycardia was not found in this study. Fourth, RUA can infiltrate through the tissue of lungs and reach capillaries and circulating cells. These RUA are then translocated to systemic organs including heart. The deposition can induce the oxidative stress and then cause the direct tissue injuries and inflammation reaction. Next, although proarrhythmic effects of RUA were mediated by oxidative stress and CaMKII phosphorylation, this study did not present the threshold level for the change of oxidative stress and CaMKII activation. Finally, it is known that UFP, unlike PM2.5, is less homogenous and influenced by the within-city sources mainly traffic. Soot particles are one of the major components of PM in the atmosphere, which are categorized into PM10, PM2.5, or PM0.1, mainly by their aerodynamic size in micrometer scale. It has been known that fine dust (PM2.5) and ultrafine dust (PM0.1) in busy cities consist mostly of soot particles and tire dust from transportation-related sources. Therefore, we experimented with a soot generator that produces PM similar to RUA. Because the main goal of this study was to determine the threshold level to induce arrhythmia, we used same soot particles and escalated the level of RUA. In current technology in our research team, it was not possible to control the level of different composition of RUA. In addition, first, the control and 800 µg/m group were performed first, and then the 200 µg/m and 400 µg/m were conducted. O2 and CO2 do not significantly deviate from the range of the control group, and temperature and humidity are conditions that do not affect the experiment. In the future, the analysis using various component according to geographical, seasonal, and temporal factors are needed.
In conclusion, RUA group could induce electrophysiological changes such as APD and QT prolongation, fibrosis, and inflammation dose-dependently, with significant increase of ventricular arrhythmia at RUA concentration of 400 µg/m3. RUA concentration of 800 µg/m3 increased phosphorylation of CaMKII activation and PLB.
 XML Download
XML Download