

 PDF
PDF Citation
Citation Print
Print



DIABETIC NEUROPATHY
The International Diabetes Federation reported that in 2019 there were an estimated 463 million adults worldwide living with diabetes, a number that projected to rise to 700 million by 2045 [1]. Of these, over half are likely to develop damage to the somatic and/or autonomic peripheral nervous system (PNS) [2]. It is also becoming increasingly appreciated that diabetes damages both the spinal cord (myelopathy) and brain (encephalopathy) such that diabetes is recognized as a prominent risk factor for developing cognitive dysfunction and Alzheimer’s disease [3]. There is no Food and Drug Administration (FDA)-approved disease modifying treatment for preventing or slowing progression of diabetic neuropathy other than the recommendation to maintain glycemic control [4] with management of symptomatic consequences such as pain [5]. Maintaining consistent normoglycemia is a challenge for most diabetic patients and while this approach may slow onset and progression of neuropathy in type 1 diabetes mellitus (T1DM) [4] it is increasingly viewed as ineffective for treating neuropathy in type 2 diabetes mellitus (T2DM) [6], which represents the vast majority of diabetic patients. There is thus an urgent need for effective therapies that prevent or reverse diabetic neuropathy. Development of such treatments has been pursued for over 50 years but has been impeded by the complex mechanisms that cause neuropathy and difficulties in translating plausible therapies from animal models to humans [7,8]. Recent advances in unraveling pathogenic mechanisms [9,10] and enhancing clinical trial design [11] have revitalized the potential for agents developed in preclinical studies to translate into novel therapeutics and also highlighted the need for improved and concordant outcome measures in preclinical and clinical research.
CLINICAL ASSESSMENT OF DIABETIC NEUROPATHY
Diabetic neuropathy most commonly presents as a distal symmetrical polyneuropathy with degeneration of both sensory and motor fibers of the PNS. People with diabetes, pre-diabetes or metabolic syndrome may first become aware of the onset of peripheral neuropathy via indicators of sensory dysfunction such as paresthesias and pain. Neuropathy progresses to sensory loss, foot ulceration and amputation and contributes to comorbidities such as fall-related injuries [12]. Recent American Diabetes Association recommendations suggest yearly screening using temperature and/or pinprick sensation for small fibers, a 128 Hz tuning fork for vibration perception by large fibers and the 10 g monofilament for risk of ulceration [13]. Such tests detect both loss and gain of function. Beyond such basic screening tests, nerve conduction studies (NCSs) are commonly used to measure large motor and sensory fiber function and draw broader inferences about progression of neuropathy and neuropathology [14]. Impact on diabetes-induced nerve conduction velocity (NCV) slowing remains the current gold standard for determining drug efficacy in clinical trials [4,11]. Quantitative sensory testing (QST) devices that measure temperature and vibration perception were developed for use in clinical trials along with questionnaires to quantify pain and quality of life [12] and there are a number of clinical scoring systems that incorporate these tests, each weighted in a different manner [15]. There is no diagnostic requirement for demonstration of nerve pathology and use of sural nerve biopsy, which was used historically to identify pathology in the nerve trunk and as an outcome measure in clinical trials [16], is not well tolerated. However, there is a growing appreciation that quantification of small fiber pathology by skin biopsy [17] and/or corneal confocal microscopy (CCM) [18] also provides sensitive measures of neuropathy and response to therapeutic intervention [18–23].
Electrophysiology
Nerve conduction studies
NCSs allow measurement of myelinated sensory and motor nerve function by transcutaneous stimulation of nerves of the arms and legs with recording of evoked sensory nerve action potentials (SNAPs) and compound action potentials in associated muscles (CMAP) via surface electrodes [24]. Comparing action potential amplitude and velocity to local normative ranges corrected for age, sex and height allows inferences to be drawn about pathophysiological conditions such as demyelination and axon loss [25]. NCS are frequently supplemented by electromyography (EMG), which requires use of needle electrodes inserted into the muscle but provides additional information to assist diagnosis. It should be noted that many electrophysiological studies in rodents use EMG to calculate NCV (see below).
Both reduced SNAP and slowing of motor and sensory nerve conduction velocities (MNCV and SNCV) are detected in patients with diabetic neuropathy, implicating both axonal loss and demyelination as underlying pathologies [25]. NCS can be used in diagnosis, staging and monitoring of progression of diabetic peripheral neuropathy and may also allow prediction of associated comorbidities such as foot ulceration and even mortality [26]. Prominent use of NCS has emerged from FDA guidance indicating that prevention, reversal or attenuation of NCV slowing serve as the primary indicator of efficacy in clinical trials of novel therapeutic agents for diabetic neuropathy. NCV slowing historically exhibited a modest rate of progression (<2% per year) in cohorts of diabetic subjects [27] and in recent years this rate may have further slowed due to general improvements in glycemic control and increased use of drugs such as ACE inhibitors and lipid-lowering agents for other complications of diabetes but which also impact neuropathy [28,29]. Consequently, use of NCV as a primary outcome measure imposes clinical trial designs requiring large cohorts of subjects with mild-moderate neuropathy who are followed over long periods of time by skilled reference labs in order to detect treatment efficacy [11,30]. This makes clinical trials extremely expensive to undertake. Moreover, NCS assess only large fiber function while there is an emerging appreciation that small fibers are damaged early in the course of diabetes and mediate the symptoms of most concern to patients [31]. Thus, while currently considered the gold standard for diagnosis of diabetic peripheral neuropathy, NCS are usually only recommended in general practice to exclude other neuropathic diseases when a diabetic patient has an atypical pattern of symptoms or signs and there are growing calls for measures of small fiber neuropathy to have equal status as outcome measures in clinical trials [19].
Rate dependent depression
A simple modification to the EMG procedures described above can be used to provide additional information on the function of spinal inhibitory systems. Adjusting the nerve stimulator to provide trains of stimuli at the voltage that gives maximal H wave amplitude allows measurement of rate dependent depression (RDD) of the H wave, in which H wave amplitude declines over consecutive stimuli [32]. Patients with spinal injury exhibit loss of this RDD [33], as do animals with loss of spinal GABAergic systems due to ischemic trauma [34]. Reports that diabetic rodents also show an absence of RDD that parallels indices of painful neuropathy (see below) have led to the hypothesis that RDD acts as a biomarker of spinal inhibitory function and can identify pain states arising from spinal disinhibition [35,36]. In support of this, a recent clinical study identified impaired RDD in a sub-population of T1DM subjects with painful neuropathy [37], offering the possibility of using this test to stratify patient entry to clinical trials of anti-pain agents with known sites of action and for personalized, lesion-specific, treatment strategies. RDD may serve as a counterpoint to microneurography, which has been used as a means of identifying subjects with potentially peripherally driven pain (see below).
Microneurography
Microneurography is used to measure electrical activity of peripheral sensory afferents via an intraneural microelectrode inserted through the skin into a peripheral nerve in the awake subject [38]. Baseline spontaneous activity may be recorded, while cutaneously-applied stimuli are used to activate individual fibers, thereby defining their sensory modality sub-type. Abnormal spontaneous action potentials in C-nociceptors have been associated with spontaneous pain [39] and increased stimulus evoked firing activity with nociceptor sensitization [40]. At present, only a limited number of studies have employed microneurography in diabetic subjects because of the invasive and time consuming protocols required by skilled investigators. It has been suggested that analgesic efficacy in human clinical trials could be accessed by microneurography [41] and the technique has been used to identify subjects with hyperactive peripheral nerves and pain for entry into a clinical trials of a therapy that specifically targeted peripheral nerve activity in order to increase potential for success [38,42]. Future studies that use both microneurography and RDD assessment (see above) could offer a powerful approach to stratifying subjects to targeted therapies.
Sensory perception and pain
Quantitative sensory testing
QST is used to detect small and large fiber dysfunction in early stage diabetic peripheral neuropathy [43]. QST involves measuring thermal threshold for subjective cold (A-δ fibers) and warm (c fibers) sensations, and may detect positive (hyperalgesia) and negative (hypoalgesia) aspects of small fiber neuropathy that NCSs cannot measure. Vibration perception by large myelinated sensory fibers, assessed using a biothesiometer, is also a component of QST and a useful predictor for foot ulceration. These standardized measures of thermal and vibration perception thresholds have been described as sufficiently reproducible to be used as efficacy endpoints in clinical trials of potential therapeutics for diabetic neuropathy [44–46], although concerns have been raised regarding use as the sole primary endpoint [47] and diagnostic efficiency compared to other tests [48]. In a recent clinical trial of C-peptide to treat neuropathy in T1DM subjects, significant efficacy was achieved using vibration perception as the indicator whereas no significant effect was noted on nerve conduction, in part due to an unanticipated placebo effect [49]. The perceived failure of this clinical trial illustrates the dominant reliance on electrophysiology in the current clinical trials climate.
Scoring of subjective symptoms
In the diagnosis of diabetic neuropathy, the subjective assessment and quantification of neuropathic symptoms and pain can be achieved by use of one or more questionnaire-based scoring systems. These questionnaires are reproducible and include standardized and validated scores for both the severity of symptoms and the degree of neuropathic deficits. Examples in current use that focus on pain include the McGill Pain Questionnaire [50], the Brief Pain Inventory [51], and the Neuropathic Pain Symptom Inventory [52]. The Michigan Neuropathy Screening Instrument [53] is also a useful tool in screening for diabetic neuropathy. Each system has its own components and weighting. For example, the Neuropathy Symptom Score includes burning, numbness, or tingling, fatigue, cramping, aching, or nocturnal exacerbation while the Neuropathy Disability Score and Neuropathy Impairment Score are calculated using ankle reflex, pinprick sensation, temperature sensation, and vibration. Based on the Toronto Clinical Neuropathy Score [54], diabetic patients can be categorized as having mild, moderate or severe neuropathy by summation of symptom scores. Repeated deployment of a battery of these questionnaires allows iterative monitoring of neuropathic symptoms and therapeutic responses throughout clinical trials. Despite reflecting the patients perspective they tend to serve as adjuncts to physiological assays of nerve function due to perceived subjectivity and variability.
Nerve pathology
Nerve biopsy
Nerve biopsy is not required for diagnosis of diabetic neuropathy given its relatively invasive nature compared to assessment of symptoms and electrophysiology as described above. Early autopsy studies provided insight into the structural damage caused by chronic diabetes, such as Wallerian degeneration and segmental demyelination but tended to emphasize late stage pathology. Biopsy studies conducted as part of research programs have allowed documentation of earlier stages of diabetes-induced neuropathology, such as microangiopathy [55] and early reactive, degenerative and proliferative changes in Schwann cells [56]. Such studies helped guide formulation of potential pathogenic mechanisms, were used to investigate potential associations between pain and nerve degeneration/regeneration profile [57] and have highlighted diabetic cats as the preclinical model that most closely reflects human diabetic neuropathology [58]. Nerve biopsies have also been used in clinical trials to assess efficacy of potential therapeutic agents to promote axon regeneration [59,60] and remyelination [16]. However, expense, assay variability and a focus on large myelinated fibers, as well as the invasive and non-iterative nature of the procedure, has limited use of nerve biopsies in clinical trials and there has been a move towards less invasive or non-invasive approaches to visualizing small fiber pathology such as skin biopsy and CCM to complement large fiber NCV slowing in clinical trials [19].
Skin biopsy
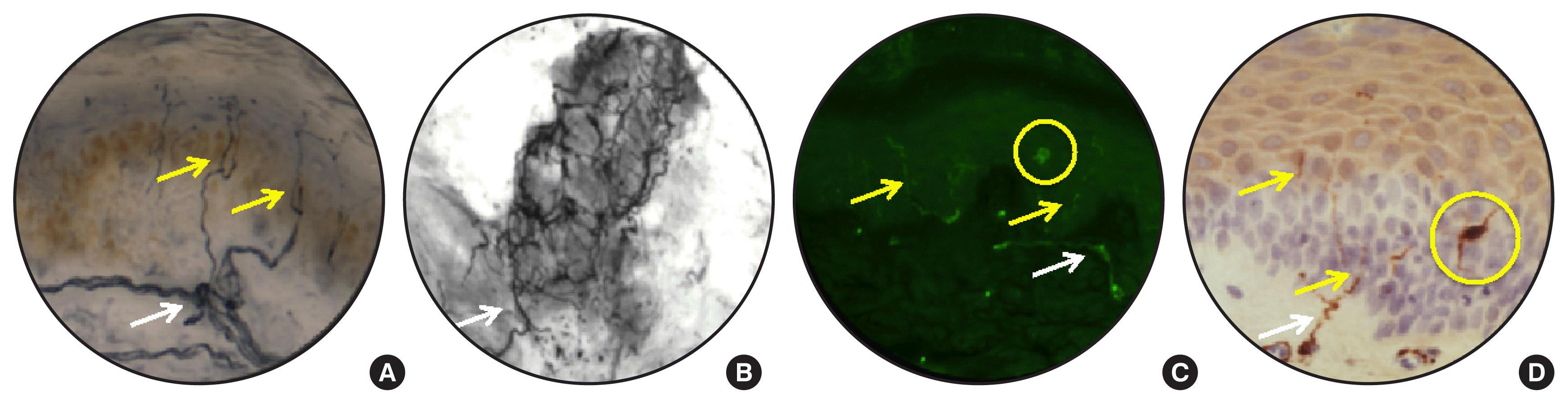
It is widely held that small sensory nerves are the first to show impairment in diabetes [61] and patients are certainly likely to have a greater awareness of sensory, rather than motor, dysfunction. Other then the sensory perception tests described above, visualization of small unmyelinated sensory fibers in the epidermis via a skin biopsy offers the opportunity to quantify distal nerve endings (intra-epidermal nerve fibers [IENFs]) as an index of small fiber pathology [17,62,63]. The standardized procedure requires a 3-mm punch skin biopsy at the distal leg and quantification of the linear density of nerves crossing the dermal:epidermal interface in at least three 50 μm thick sections per biopsy. Mean dendritic length, a measure of all nerve length in the epidermis can also be quantified to assess early remodeling of IENF prior to retraction/degeneration or collateral sprouting in response to therapy that is independent of regrowth from the dermis [63,64]. Sensory nerves are visualized using anti-protein gene product 9.5 antibodies [65] and bright-field or immunofluorescence microscopy, with the caveat that Langerhans cells, dendritic cells of the epidermis, are also stained (Fig. 1) so that their processes must be differentiated from IENF [66]. Other than nerve density, discrete axonal swellings within IENF can also be quantified [67] and have been suggested to be linked to early pathophysiological processes of axonopathy [68–70]. Nerve regeneration capacity can also be assessed by combining local denervation by topical capsaicin with subsequent skin biopsy and IENF quantification [71]. Because skin biopsies capture the epidermis, the papillary dermis and portions of the reticular dermis, dermal myelinated nerve fibers innervating dermal mechanoreceptors and autonomic nerve fibers innervating sweat glands may also be quantified (Fig. 1) [72].
Diabetes causes progressive loss of IENF to almost complete ablation. There is early loss of IENF in subjects with impaired glucose tolerance and diabetes that precedes significant large fiber NCV slowing [73] so that it is considered to be an objective means to detect early small nerve damage in diabetes. Skin biopsies are not widely used in clinical diagnosis of diabetic neuropathy because they are invasive, slow to provide data and costly compared to other tests [17,62,70]. However, the ability to repeatedly sample adjacent biopsy sites over time means they are becoming an important component of clinical trials, particularly for agents that target mechanisms of nerve regeneration in small fibers. IENF regeneration is inherently impaired by diabetes [71] and only a few studies have reported improvement of epidermal innervation after intervention. In T2DM subjects, 10 weeks of increased aerobic exercise increased epidermal nerve fiber branching [74] while longer periods of lifestyle modification significantly increased IENF density in subjects with impaired glucose tolerance or T2DM [75,76]. Simultaneous pancreas and kidney transplant that effectively cured patients with severe T1DM also increased mean dendritic length of IENF, although not IENF quantified by the standard clinical method [64]. Assessment of IENF in skin biopsies provides a useful measure of small fiber pathology to accompany QST and large fiber NCV studies and should perhaps serve as the primary outcome measure in clinical trials of agents that target small fiber regeneration. As IENF loss also occurs in rodent models of diabetes (see below), the assay may also serve as a suitable bridge for translating experimental therapies.
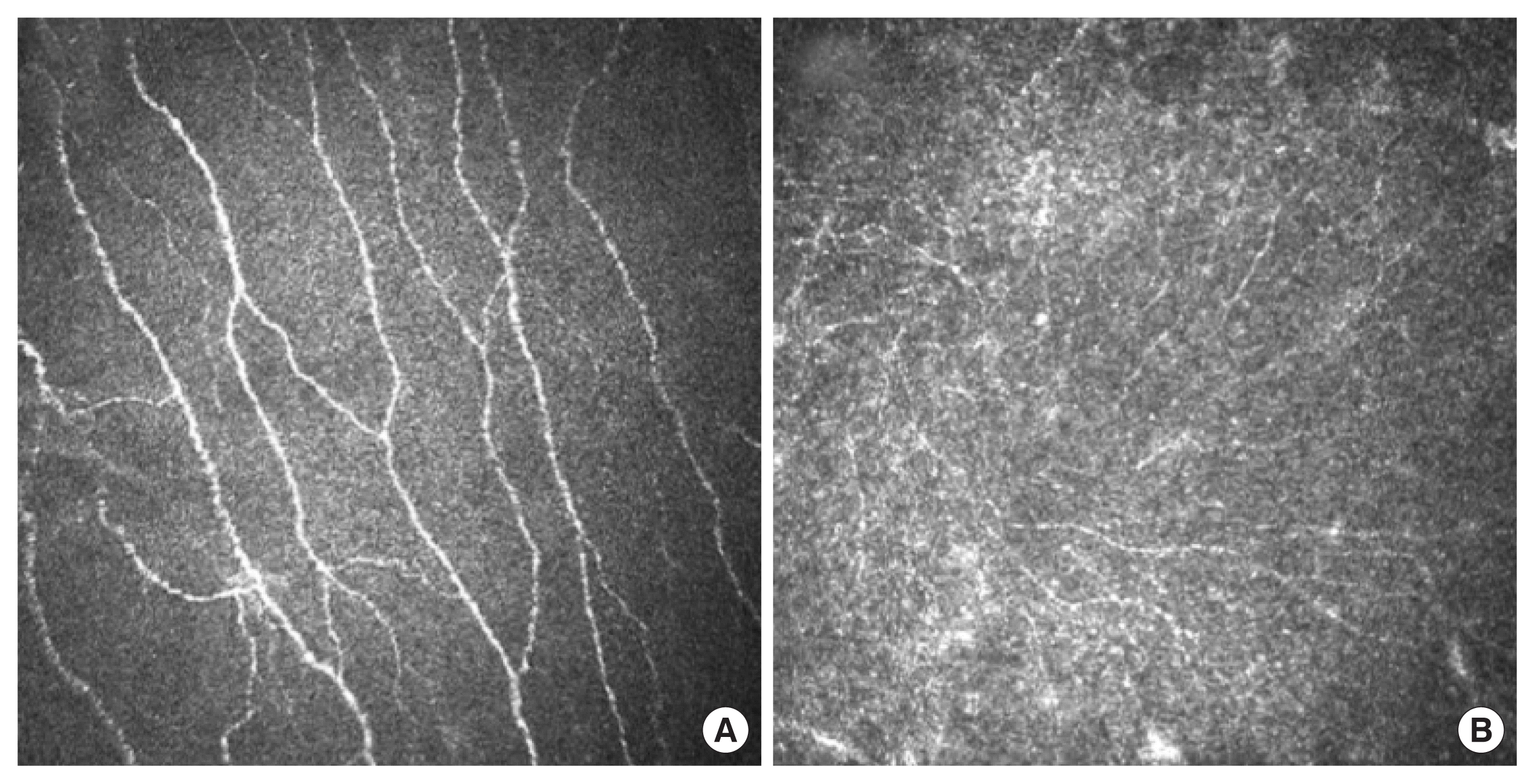
Corneal confocal microscopy
CCM is an emerging tool being developed for measuring impact of systemic diseases such as diabetes on small sensory fibers [77]. It visualizes in real-time the sub-basal nerve plexus of the cornea without need for contrast agents (Fig. 2) and is a non-invasive and relatively inexpensive procedure that allows iterative measurements of the same nerves. Automated image analysis algorithms have been developed to remove inter-observer variability and accelerate data delivery [78,79]. These features support suitability for tracking progression of neuropathy and also recovery following therapeutic intervention [80]. Loss of corneal nerve density correlates with severity of neuropathy and other measures of small fiber neuropathy such as IENF loss [22] and CCM can detect early small-fiber axonal damage in diabetic neuropathy [81,82]. The sensitivity and specificity for the diagnosis of diabetic neuropathy have been established [83] and corneal nerve loss predicts incident neuropathy in those with poorer glycemic control and lower high density lipoprotein [84]. At present there are few studies showing recovery of corneal nerve loss in diabetic subjects treated with therapeutic agents, other than one showing increased corneal nerve density following 1 year of improved glycemic control [85]. The most compelling data comes from a longitudinal study of formerly diabetic patients who underwent simultaneous pancreas and kidney transplantation that effectively cured diabetes. Significant improvement in corneal nerve parameters preceded detectable efficacy against other indices of neuropathy such as neuropathy symptom profile score, large fiber conduction slowing and reduced IENF density [64]. Despite the promising advances, there is ongoing debate regarding how well corneal nerves will serve as surrogates or biomarkers for other sensory nerves during systemic disease, given their relatively short length and unique local environment. Nevertheless, use of CCM to monitor small fiber neuropathy is an exciting development that, like IENF loss, can also be used preclinical models of diabetes.
MODELS OF DIABETIC NEUROPATHY
Many of the assays used to identify neuropathy in diabetic patients are also amenable to use in preclinical models [86] and are used to identify agents with most translational potential, provided that the disorders share the same pathogenic mechanisms.
Cell culture
Early in vitro studies using immortalized cells lines have evolved so that Schwann cells and peripheral sensory neurons derived from adult rodents are now widely used to investigate cellular mechanisms linked with diabetic neuropathy. Such studies have identified a number of plausible pathogenic cascades that warrant further exploration in more complex systems [87,88]. Advantages of using such systems include the ability to design relatively fast studies (over days–weeks, not months–years), control of environmental conditions and drug delivery, the ability to track real time intracellular events within a specific cell type and minimal ethical concerns. There is particular value in using sensory neurons derived from adult rodents to identify agents that promote neurite outgrowth and thus have potential for encouraging axonal regeneration following traumatic injury or protecting against neuropathy [89]. However, there are also caveats regarding the pertinence of excised tissues maintained in an artificial (and probably incomplete) environment, particularly as transection of axons during dorsal root ganglion (DRG) removal prompts an injury and regeneration phenotype while Schwann cells revert to a non-myelinating status. It is also worth noting that most culture media used to sustain neurons already contains glucose concentrations of 20 mM or more, this being in marked excess of those considered hyperglycemic in vivo, making experimental design and interpretation difficult. Sophisticated cell culture systems employ cells derived from adult normal and diabetic animals that are excised and maintained under in vitro conditions that reflect the source glycemic and insulin environment [90] and co-culture Schwann cells and neurons to promote myelination [91,92].
Animals
Diabetic cats develop pathology that most closely reflects advanced human diabetic neuropathy, with overt demyelination and axonal degeneration in nerve trunks [58]. However, rats and mice are most widely studied as models of diabetic neuropathy and are responsible for providing the vast majority of data that supports current hypotheses of the pathogenesis of diabetic neuropathy and translation of potential therapies. Both rats and mice can model pre-diabetes, T1DM or T2DM using genetic, chemical or dietary insults and while there are model-specific variations in the presentation and progression of neuropathy, there is a broadly consistent phenotype that models the human condition of both loss and gain of function [93].
Rodent models of T1DM or T2DM reliably show many functional disorders of the early human condition, including large fiber MNCV and SNCV slowing and allodynia/hyperalgesia to sensory stimuli that in some cases, such as heat hyperalgesia in diabetic rats, progresses to sensory loss [86]. They also go on to replicate some features of early structural pathology in humans such as reduced density of epidermal [94] and corneal [95] small sensory nerve fibers, along with myelin thinning and reduced caliber of large myelinated sensory and motor axons and their perikarya [96–100]. Descriptions of encephalopathy with accompanying cognitive impairment [101, 102] suggest viability as models of the Alzheimer’s Disease-like condition increasingly reported in long-term diabetes [103].
Aside from inescapable features of rodent biology such as short life span, physically short nerves and tolerance of levels of hyperglycemia (25 to 60 mM) that are fatal in humans, there are also a number of other caveats to be borne in mind when using rodent models of diabetic neuropathy or interpreting data from such studies. One major weakness of rodent models is their poor replication of the late structural pathology of human diabetic neuropathy. Most intriguingly, there is little overt Schwann cell pathology or demyelination, despite this being a prominent feature of the human condition. After half a century of neuropathologists failing to find reliable evidence of overt myelin disruption in rodent models of diabetes, other than mild myelin thinning detected in nerve cross sections and occasional segmental demyelination and reduced internodal distance measured in teased fibers [96,104–109], there is a current tendency to identify dramatic myelin damage within weeks of onset of diabetes in the same models. Whether this reflects evolution of the models to a more desirable phenotype or enhanced/diminished interpretative skills in the research community will presumably be resolved over time. Features indicative of demyelination and subsequent remyelination such as thin myelin with supernumerary Schwann cells can be reproduced by superimposing hypertension onto T1DM rats [110]. Hypertension is a major risk factor for neuropathy in diabetes [111] and, as many rodent models of diabetic neuropathy are normotensive or mildly hypotensive [110], a lack of cumulative risk factors may restrict the rapid evolution of a human-like neuropathology phenotype.
Concerns have been raised that the pancreatic beta cell toxin streptozotocin (STZ), which is widely used to produce rodent models of T1DM, causes direct neurotoxicity. These concerns arose initially when STZ-injected animals that did not develop hyperglycemia showed indices of neuropathy [112]. Subsequent studies associated mechanical hyperalgesia with STZ-induced insulin depletion that, while not sufficient to induce overt hyperglycemia, presumably impacted insulin signaling in nerve [113,114]. Thermal hyperalgesia was also linked to increased expression of the transient receptor potential vanilloid receptor 1 (TRPV1) channel via oxidative stress [115]. More recently, STZ has been reported to directly activate transient receptor potential ankyrin subtype 1 (TRPA1) receptors on adult-derived sensory neurons in vitro and produce an acute, transient allodynia following intraplantar injection in mice that resolved within 24 hours–before onset of diabetes [116]. However, in vivo STZ is largely removed from the blood within 15 minutes and cleared in the urine within 2 to 6 hours [117]. Initial (2 to 3 days) hyperinsulinemia and hypoglycemia due to pancreatic beta cell necrosis is followed by hypoinsulinemia and hyperglycemia. Most indices of neuropathy emerge after 2 to 8 weeks of hyperglycemia and appear unlikely to reflect direct neurotoxicity of STZ. Nevertheless, such concerns highlight the need to establish the provenance of newly-identified disorders in STZ-diabetic rodents which can be achieved using 3-O-methyl glucose [37,118], by reversal of established disorders with insulin therapy to continuously normalize blood glucose levels [37] and parallel use of models of diabetes arising from genetic or dietary insults [119].
The widely used rodent models of diabetes are perhaps best viewed as modeling early functional and structural indices of neuropathy. This allows molecular and biochemical changes in nerve to be interpreted as potential pathogenic mechanisms that lead to degenerative neuropathy. However, it should be remembered that the mild neuropathy of these models prevents guarantees that any therapies emerging from their use will prevent or reverse the more advanced stages of clinical diabetic neuropathy.
Assays
Electrophysiology
Preclinical studies of NCV frequently utilize shifts in M and H wave latency of the EMG following stimulation at different sites to calculate velocity [86]. MNCV/SNCV slowing occurs within weeks of onset of diabetes in almost all animal models of diabetes and is commonly used in preclinical drug development as a primary measure of therapeutic efficacy, in part because this has been the FDA-defined primary end point in most clinical trials (see above). However, it must be cautioned that having the same dysfunction as humans does not automatically indicate that disorders measured in both species arise by the same mechanism. For example, NCV slowing in short term diabetic rodents is initially of metabolic origin and not associated with any detectable structural pathology to axons or myelin [120], whereas axonal degeneration and demyelination are prominent in humans [55] and likely to contribute to NCV slowing in subjects enrolled in clinical trials [11]. This discord may explain why the impressive efficacy of drugs that interrupt glucose metabolism against NCV slowing in short term diabetic rodents [121] has not been replicated in clinical trials, where only modest effects have been reported [30,122].
CMAP is relatively stable in humans due to the large muscle groups being sampled, thereby allowing inferences about axonal and muscle pathology. In contrast, CMAP is much less stable in rodents, so that use of this aspect of clinical electrodiagnostic testing to assess between-animal and between group pathology is less reliable. Nevertheless, measuring CMAP amplitude during bursts of stimulation to a restrained limb can replicate the clinical observation that maximal M wave amplitude remains constant while maximal H wave amplitude in the same EMG declines in a frequency-dependent manner [32]. RDD in rodents is attributed to release of GABA as the initial stimulus traverses the spinal cord with inhibition of subsequent components of the stimulus train via actions on spinal GABAA receptors [35]. RDD thus serves as an electrical biomarker of spinal GABAergic inhibitory functions. RDD is impaired in diabetic rodents due to disruption of the GABAA receptor inhibitory functions secondary to decreased expression of the potassium and chloride transporting potassium-chloride cotransporter 2 (KCC2) pump [35] located in the post-synaptic membrane. As both RDD and indices of neuropathic pain covary in diabetic rodents and both respond in a similar manner to assorted spinal pharmacological manipulations it has been proposed that loss of RDD may serve as a biomarker for pain in which loss of spinal GABAergic inhibition plays a role [36]. Potential clinical utility of this phenomenon is suggested by a recent clinical study indicating that a sub-group of T1DM patients with pain show loss of RDD [37].
Sensory perception and pain
Behavioral responses to sensory stimuli are widely used as surrogates of presumed pain perception in diabetic rodents, with all the attendant caveats that apply to use of such evoked response tests when used to define other models of neuropathic pain [123,124]. The physiology of diabetes brings additional concerns that muscle wasting and protein glycation in skin can also modify behavioral responses. Commonly reported indices of painful neuropathy in multiple rodent models include allodynia to von Frey filaments, hyperalgesia in the paw pressure (Randall-Siletto), hot plate and Hargreaves heat tests (with progression to hypoalgesia in some longitudinal studies), allodynia/hyperalgesia in the acetone and cold plate tests and allodynia/hyperalgesia in the paw formalin test [125]. Not all sensory tests are universally disrupted in all models of diabetes. Anomalies include hyperalgesia in the paw formalin test of STZ-diabetic rats [126] but hypoalgesia in STZ-diabetic mice [127] and the persistence of heat hyperalgesia in some studies but progression to hypoalgesia in others. The latter may reflect relative severity of STZ-induced insulin depletion and unreported use of trace insulin to protect against extreme cachexia, as trace insulin therapy that does not impact hyperglycemia prevented the progression from heat hyperalgesia to hypoalgesia [128]. Increased sensitivity of diabetic rodents to von Frey filaments appears to be particularly reproducible between species, strains, models and investigators and is widely used to evaluate potential analgesics despite brush-evoked tactile allodynia not being a universal representation of painful neuropathy in diabetic subjects [129].
Diabetic rodents do not display behaviors associated with extreme or spontaneous ongoing pain such as autotomy, guarding behavior, vocalization upon touching the body or ultrasonic vocalizations [130], and well-maintained diabetic rodents are indistinguishable from controls in open-field behavior. Evidence for spontaneous pain in diabetic rodents has been largely restricted to electrophysiological studies suggesting spontaneous or enhanced evoked activity in peripheral nerve [131,132] and/or spinal cord [133–136] and bears the caveat that stimulus-evoked neurotransmitter release in the cord of diabetic rats is reduced, not increased [137,138]. Conditioned place preference (CPP) tests have been deployed to assess use of anti-pain drugs and employ preference as a surrogate for ongoing pain. Initial studies in STZ-diabetic rats disassociated discernable effects of a TRPA1 channel antagonist on mechanical hyperalgesia from lack of effect in a CPP assay and therefore were unable to support the implication of ongoing pain in the model [139]. More recent studies demonstrated CPP using soluble epoxide hydrolase inhibitors [140,141] or pregabalin [142] that also alleviated abnormal responses to evoked pain tests. Such findings are likely to support addition of CPP to the battery of tests viable in diabetic rodents for use in drug development programs.
Nerve pathology
As discussed above, degeneration of nerve fibers in peripheral nerves of diabetic rodents is controversial, with many careful morphometric studies suggesting that pathology during the first few months of diabetes is limited to reduced perikaryal and axonal diameter and eventual myelin thinning in the absence of overt demyelination, degeneration, and axonal loss. These features have the potential to contribute a structural component to large fiber conduction slowing in long-term diabetic rodents. Reports of myelin disruption and frank demyelination in short term diabetic rodents have yet to be supported by convincing documentation using electron microscopy. Like humans, small sensory fibers may be visualized in skin biopsies and in situ via CCM (Figs. 1 and 2) and diabetes causes a progressive reduction of small fiber density in both tissues [95,143]. The functional consequences may be reflected in paw thermal hypoalgesia and diminished corneal reflexes. However, onset of hypoalgesia precedes detectable loss of epidermal nerves in diabetic mice [144] and early neurochemical disorders such as impaired synthesis, transport and evoked spinal release of sensory neurotransmitters [137,138,145,146] may also contribute to early sensory loss. Whether loss of corneal and epidermal sensory nerves in diabetic rodents shares the same pathogenesis as that seen in humans remains to be established.
CONCLUSIONS AND SPECULATION
A major value of preclinical models of disease comes from identification and stress testing of potential pathogenic mechanisms and therapeutic approaches. The capacity to translate preclinical discoveries into therapeutic approaches for human disease is largely dependent on the fidelity of the preclinical models to the human condition and use of common assays of therapeutic efficacy. Unfortunately, the translation from preclinical promise to clinical approval for therapies directed at diabetic neuropathy over the last 50 years has been littered with failure, with the blame equally distributed between imperfect drug design, preclinical models and clinical trial design. The historical focus on amelioration of NCV slowing in short term diabetic rodents as the primary readout to predict efficacy against NCV slowing in clinical trials may have contributed to these translational failures, given the largely metabolic dysfunction underlying the rapid and dramatic NCV slowing in rodents and the substantial structural contribution to the slowly evolving NCV slowing in humans [147]. For example, while aldose reductase inhibitors completely protect NCV in diabetic rodents [121], the minor impact reported in the most thorough clinical studies [30] may indeed reflect the best achievable outcome for a metabolic intervention.
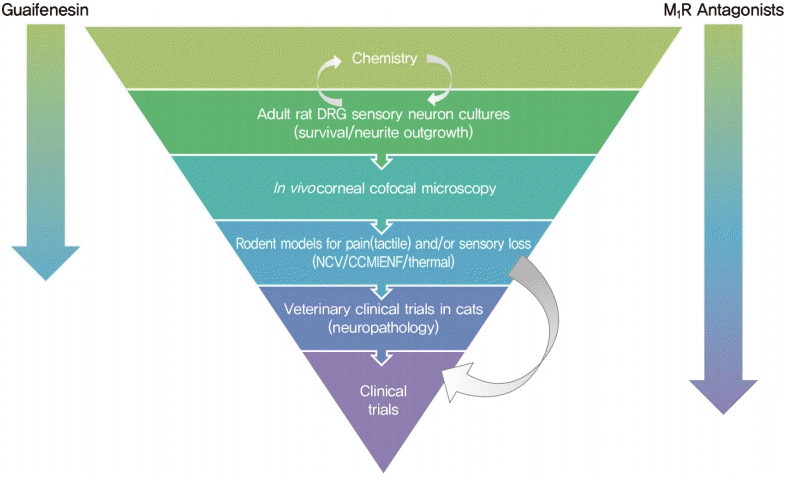
The growing appreciation that diverse pathogenic mechanisms deriving from both glucotoxic and non-glucose driven primary lesions may contribute to diabetic neuropathy [9,10] is driving both consideration of polypharmaceutical approaches [148] and also use of more refined preclinical development programs that require progression through a variety of model systems using efficacy readouts with direct clinical equivalents. One such development funnel, designed to identify therapies that promote nerve regeneration and neuroprotection, is shown in Fig. 3. Compounds emerging from synthesis platforms or existing libraries are initially screened for axonal growth and survival properties using adult sensory neuron cultures derived from normal and diabetic rodents, with hits being refined by medical chemistry approaches before progressing to assay of sensory nerve growth in the cornea of normal and diabetic rodents after topical delivery. This provides a relatively fast path to first in vivo data. Efficacy supports advancement to systemic delivery studies in diabetic rodents using multiple small and large fiber indices of neuropathy. Veterinary clinical trials in diabetic cats offer an optional late screen for impact on nerve pathology that mirrors the human condition. In one example of this approach, an in vitro screen of 600 compounds using sensory neurons derived from adult rat DRG identified guaifenesin as a potent enhancer of neurite outgrowth while subsequent structural refinements isolated the biological activity to the R-enantiomer [89]. This was unexpected, as guaifenesin is commonly included in cough medicine formulations as an expectorant but had no reported neuron-stimulating properties. Subsequent studies in diabetic mice demonstrated that both the R-enantiomer and racemic guaifenesin attenuated MNCV slowing. In another example, in vitro studies identified neuritogenic properties of non-selective muscarinic antagonists and also both selective and specific antagonists of the M1 receptor (M1R) sub-type. Varying the in vitro assay by using sensory neurons from M1R knock out or overexpressing mice confirmed the focus on this receptor sub-type, while coupling with assays of calcium signaling and mitochondrial respiration allowed the molecular and cellular consequences to be investigated [149]. Predictive strength of the in vitro system was supported by subsequent studies showing that treatment with a M1R selective antagonist prevented and reversed small fiber dysfunction, along with large fiber conduction slowing, in multiple rodent models of T1DM and T2DM and advancement to phase 2 clinical trials. While this is not the only viable testing funnel, it is hoped that use of multiple preclinical assays that replicate both functional and structural indices of human diabetic neuropathy will reduce the incidence of false positives and identify more robust therapies that translate to clinical use.
 XML Download
XML Download