

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Gut microbiome plays significant role in several cardiometabolic disorders including type 2 diabetes mellitus (T2DM) [1]. Alteration in gut microbiome composition commonly referred as dysbiosis and its consequential functional disturbances attributed to pathogenesis, insulin resistance and treatment response in diabetic individuals [23]. Recent studies have suggested that an altered gut microbiome and the metabolites derived from microbes can alter intestinal barrier, signaling pathways, modulate host physiology and metabolism that directly or indirectly contributes to diabetic states in the host [45].
In order to understand the role of microbiome on host metabolism and disease, it becomes essential to determine the functional attributes of the microbiome [6]. The metabolic capacity of the microbiome can either be inferred using 16S libraries correlating the phylogenetic trees and clusters of genes shared between taxa [7]. Alternatively, cataloged using shotgun metagenomics libraries providing a direct assessment of the functional attributes of the microbiome depending on sequencing depth [8]. Several studies have reflected on the mechanisms by which microbiome affects the host physiology, assessed from a gene content/functional perspective. For example, it is evident that gut microbiome synthesizes short chain fatty acids, including butyrate, acetate, and propionate. These have been found to be critical for enterocyte homeostasis, and their imbalance has been documented in diseases such like T2DM and inflammatory bowel disease [9]. Similarly, both inflammatory bowel disease and obesity are associated with enrichment of enzymes in the nitrate reductase pathway. Obesity also affects the metabolism of choline and p-cresol, as well as the phosphotransferase system, required for assimilation of dietary carbohydrates [10].
With the advancements in metagenomics approaches, there is a growing interest in exploring the functionality of the microbiome populations through metabolic pathways and products to better understand the interplay between microbiome and host physiology. While most studies have rallied around short chain fatty acids, enzymes, gaseous products including carbon dioxide, hydrogen, methane, and hydrogen sulfide [11], little is known about vitamins and microbiome micronutrients. Vitamin K2 (also known as menaquinone) synthesized by gut microbiome and found in meat, eggs, curd, cheese, and soybeans is known to play significant role in osteoporosis and cardiovascular diseases [1213]. There are some suggestions that vitamin K2 may be playing some role in improving insulin resistance and reducing T2DM risk through its anti-inflammatory properties, lipid-lowering effects, and the involvement of vitamin K-dependent-protein osteocalcin [1415]. However, the precise link between gut microbiome composition, involved metabolic pathways and vitamin K2 synthesis in T2DM remains unknown. In this study, we explored the metabolic capabilities of the gut microbiome among T2DM and healthy individuals through shotgun metagenomics using MiSeq platform (Illumina, San Diego, CA, USA).
METHODS
Ethical statement
Ethical approval was obtained from University Hospital Sharjah-Hospital Ethics Research Committee (UHS-HERC-021-07022018). All subjects were recruited at University Hospital Sharjah (Sharjah, UAE) and written informed consent was obtained.
Stool sample collection and preparation
We collected 18 stool specimens from adult Emirati citizens. Of the 18 subjects, 12 were T2DM and six were healthy subjects. We recruited patient chaperons accompanying T2DM patients during their hospital visits, labeled as healthy control. We determined their health status of controls using a brief medical history, vital signs, and no existing co-morbidities such as T2DM. The basic demographic information such as age, gender, marital status, education, diet, exercise, height, and weight were documented. Glycosylated hemoglobin (HbA1c) measurements were recorded from the patient electronic files. From each subject, 2 to 4 g of freshly passed stool specimen was collected in a sterile container stored immediately into liquid nitrogen and then transferred to −80℃ for further analysis. Liquid (diarrheal) stool and use of antibiotics or probiotics in the last 3 months among the volunteers were the only exclusion criteria used in this study.
Experimental design
In order to detect differences in gene potential between healthy and diabetic subjects we sub stratified the samples based on their diets (high and low fiber) calculated using dietary fiber intake short food frequency questionnaire (DFI-FFQ) [16], and HbA1c (marker of average level of blood sugar over the past 3 months) data as shown in Supplementary Table 1.
DNA extraction and library preparation
Faecal samples were subjected to DNA extraction using QIAamp PowerFecal DNA kit (Qiagen Ltd., Hilden, Germany) following the manufacturer's instruction. DNA quality was evaluated visually via gel electrophoresis and quantified using a Qubit 3.0 fluorometer (Thermo-Fischer, Waltham, MA, USA). Libraries were prepared using an Illumina Nextera library preparation kit following the standard protocol.
Sequence technology and processing
Sequencing was done using an Illumina NextSeq. Around 31.6 Gbases were generated using 2×150 paired-end reads. Each sample yielded a median of 1.8 Gbases. After sequencing, reads were separated according to the barcode used in the library preparation. Initial quality evaluation was done using FastQC v0.11.5. Processing took part in three steps: Paired-ends read joining, removing of contaminants, and trimming. Paired-end reads were joined using FLASH v1.2.11 [17]. Reads were then compared to the Human Genome (hg19, GRCh37 Genome Reference Consortium Human Reference 37) and sequences that mapped to it were removed. Finally sequences were trimmed according to their quality values using Trimmomatic v0.36 [18] using custom parameters (LEADING:5 TRAILING: 5 SLIDINGWINDOW:4:15 MINLEN:70). Joining the paired reads reduced the library size in average by 41.78%. A 0.06% of the stitched reads mapped to the human genome and then removed.
Statistical analysis
To evaluate beta diversity across samples, we excluded operational taxonomic units (OTUs) occurring in fewer than 10% of the samples with a count of less than three and calculated Bray-Curtis indices. We tested beta diversity, underscoring differences across samples, using non-metric multidimensional scaling (NMDS) ordination. Dissimilarity in community structure assessed with permutational multivariate analyses of variance (PERMANOVA) with treatment group as the main fixed factor and using 4,999 permutations for significance testing. All analyses conducted in the R environment.
RESULTS
Metagenomic characterization of diabetic and healthy microbiome
In this study, we conducted a metagenomics characterization of 18 individuals and total bacterial DNA extracted from corresponding fecal samples, then subjected to Illumina shotgun sequencing. Recovered read pools ranged from 14,554,254 to 6,945,034. At the end of quality control, median number of quality-filtered reads per sample was 10,164,901 visualized in Supplementary Fig. 1. These data then used to generate metabolic pathway profiles in the analyzed microbiome utilizing a custom script based on the MetaCyc database [19]. Clinical characteristics of diabetic and healthy individuals were also documented. In order to identify microbial biomarkers mainly attributed to T2DM, we selected individuals with uniform age, gender, ethnicity, diet, and BMI (Supplementary Table 2). Taxonomic composition was determined using Metaphlan2 [20]. The community dominated by bacteria, which accounted in average for 99.051% of the total community followed by archaea, which accounted for 0.863%. On the bacterial side, Firmicutes, Bacteroidetes, and Actinobacteria, which accounted on average for 42.54%, 27.02%, and 20.43% of the bacterial community respectively, dominated the samples. The most common Eukaryotes was Saccharomyces cerevisiae, which accounted for 1e-04% (Supplementary Fig. 2). Firmicutes and Bacteroidetes-dominated microbiome of the adults in this study is consistent with and representative of examined microbiome metabolic profiles in previous reports (Supplementary Fig. 3) [2122].
Community composition: visualizing similarity among microbiome
In order to visualize similarity among microbiome, we examined community compositions. To obtain a graphical representation of similarity among microbial communities, we used an ordination approach. The similarity between any two samples, according to their microbiome composition calculated using Bray-Curtis dissimilarities, a measure that considers both presence/absence and abundance of the species. The distances then evaluated and represented graphically using NMDS ordination (Fig. 1).
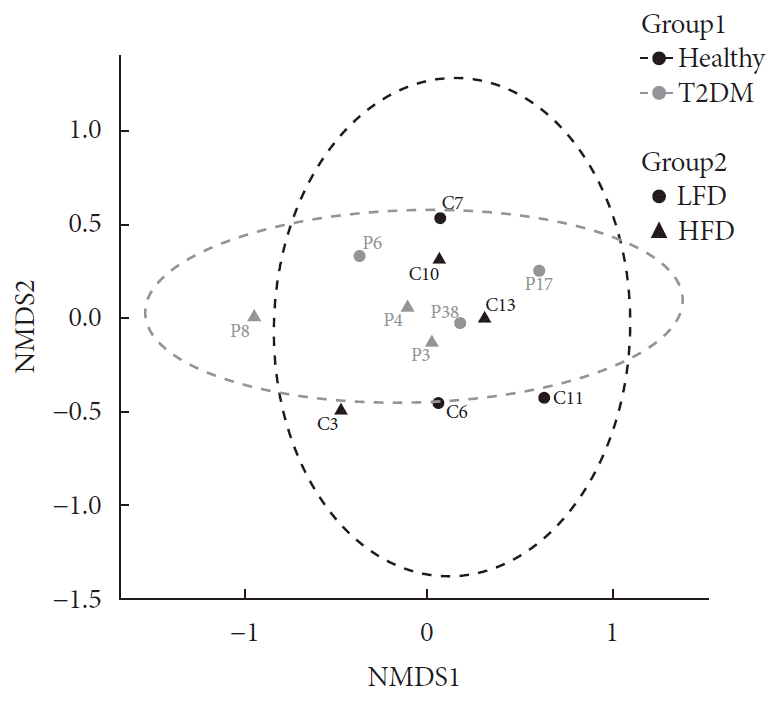
Fig. 1
Evaluation of community composition among samples. Panel shows non-metric multidimensional scaling (NMDS) ordination. Ordination plots were categorized by color according to the patient health status (Group 1) and by shape according to their diet (Group 2). T2DM, type 2 diabetes mellitus; LFD, low fiber diet; HFD, high fiber diet.
![]()
Based on PERMANOVA (using the adonis R function) determined the significance of differences between Group 1 and Group 2, none of the experimental factors (Group 1 and Group 2) or their interaction explained significantly the variation of taxonomic composition profiles. Next, differential abundance testing using DESeq2 (R package) evaluated the distribution of bacterial species between health status when controlling for differences in diet. No bacterial species found to be differentially abundant between the healthy and diabetic patients. Likewise, there was no difference in bacterial species abundance in T2DM with high HbA1c (>6.5%) and low HbA1c (<6.5%), data not shown.
Functional profiles
To elucidate functional diversity among groups, we summarized functional profiles into pathways using the Metacyc pathway definition [19]. Around 63.54% reads then mapped into functional genes. Difference in pathway richness (number of unique pathways) then calculated using a generalized linear model assuming and deemed significant. The health status was the only factor that explained significant (P<0.001) differences in richness among patients. This is important to highlight as we normalized for diet as explained before in Supplementary Table 1. Diabetic patients had higher richness of functional genes than healthy subjects did (Fig. 2).
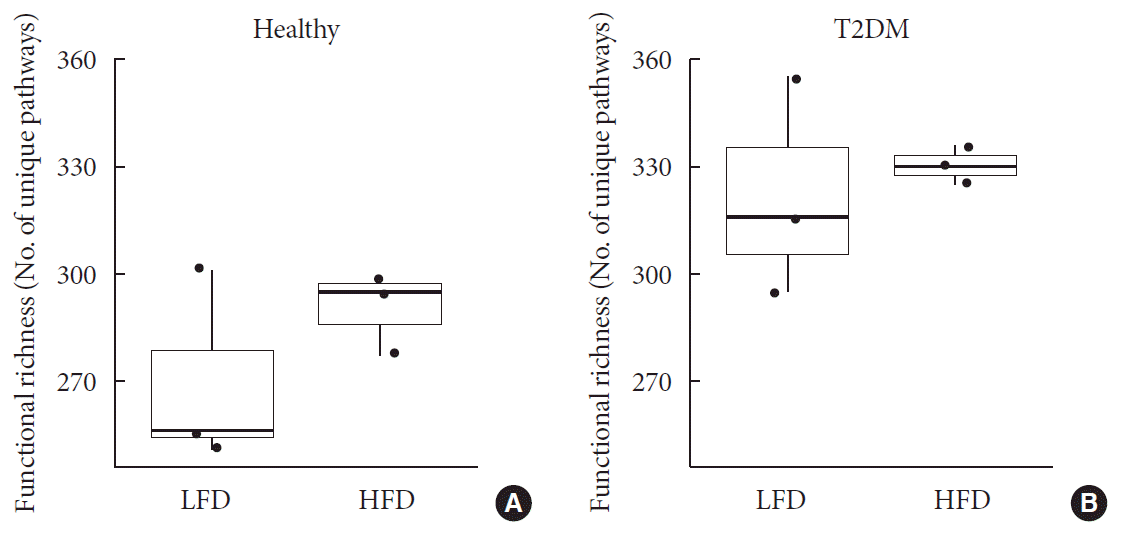
Fig. 2
Difference of functional profiles among samples. Panel shows difference in pathway richness (number of unique pathways) for (A) healthy and (B) type 2 diabetes mellitus (T2DM). Diabetic patients had higher richness of functional genes than that of healthy subjects (P<0.001) irrespective to differences in diet. LFD, low fiber diet; HFD, high fiber diet.
![]()
Furthermore, we analyzed variation of functional groups using PERMANOVA (using the adonis R function) to determine the significance of differences between healthy and T2DM. Interestingly and in contrast to the taxonomic profiles similarity in Fig. 1, one experimental factor (T2DM vs. healthy status) explained significantly part of the variation in functional gene profiles at approximately 21%.
Differential abundance testing of functional groups
Metagenome-wide association studies (MGWAS) developed to characterize gut microbiome in human disease, including T2DM and identified biomarkers that can help predict disease occurrence and possibly management outcomes [2324]. Here, we conducted a differential abundance testing of identified functional groups between T2DM, T2DM with high HbA1c, and healthy individuals. Differential abundance testing using DESeq2 (R package) and functional profiles using the Metacyc pathway, summarized into three pathways that significantly differed among healthy, diabetic, and diabetic with high levels of HbA1c subjects (when controlling for differences in diet; P<0.001) (Fig. 3). The table shows log-2-fold differences when compared the differentially abundant pathways between healthy, diabetic, and diabetic with high HbA1c subjects. These pathways are super pathway of menaquinol-8 biosynthesis I (vitamin K2) (PWY-5838) at 30 log-2-fold differences, L-lysine biosynthesis II (lysine acetylation) (PWY-2941) at approximately one log-2-fold differences, and creatinine degradation I at approximately one log-2-fold differences (Fig. 3B).
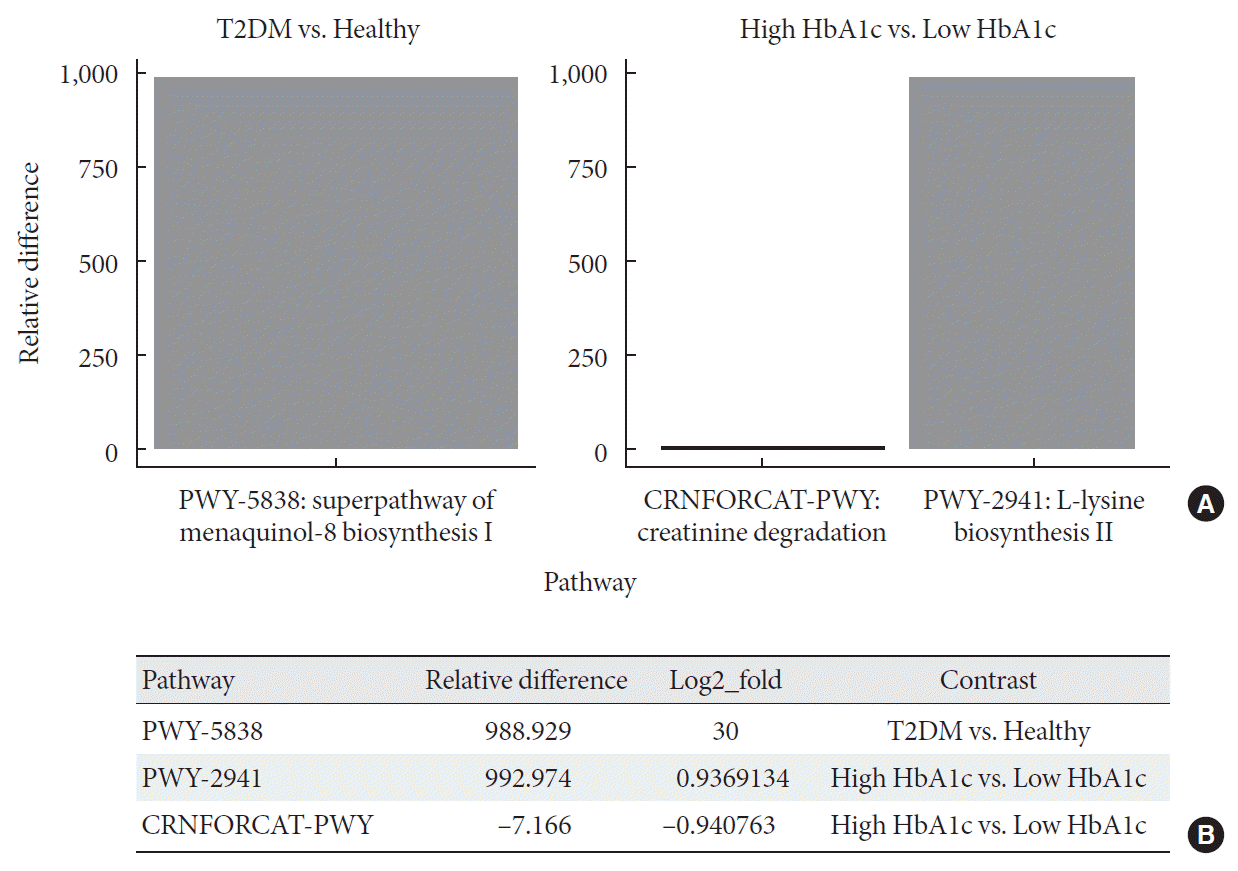
Fig. 3
Differential abundance testing of functional groups. (A) Panel shows pathways with significant relative abundance (P<0.001). (B) Log fold difference for significantly enriched pathways for type 2 diabetes mellitus (T2DM) and T2DM with high glycosylated hemoglobin (HbA1c) are PWY-5838 30 fold, PWY-2941 0.9 fold, and CRNFORCAT-PWY −0.9 fold in contrast to healthy and T2DM with low HbA1c.
![]()
Covariance network of bacterial genera and metabolic pathways
Furthermore, we evaluated covariance between the bacterial genera and metabolic pathways displaying difference in abundance (analysis of variance P<0.05) in T2DM as compared to healthy subjects. Force-driven network representation of these data revealed that genera belonging Firmicutes phylum (10 covariances, P<0.05), Actinobacteria (five covariances, P<0.05), and the Bacteroidetes (five covariances, P<0.05) utilize a key role in modulating vitamin K2 metabolic functionality of the gut microbiome that are altered during T2DM (Fig. 4). Erysipelotrichaceae belongs to Firmicutes phylum representing four out of 10 covariances correlates significantly vitamin K2 pathway (PWY-5838) enrichment in diabetic subjects (Fig. 4). Moreover, Corynebacterium is another gram-positive bacterium that belongs to Actinobacteria phylum represents three out of 10 covariances correlates significantly with vitamin K2 pathway [25] (PWY-5838) enrichment in diabetic subjects (Fig. 4).
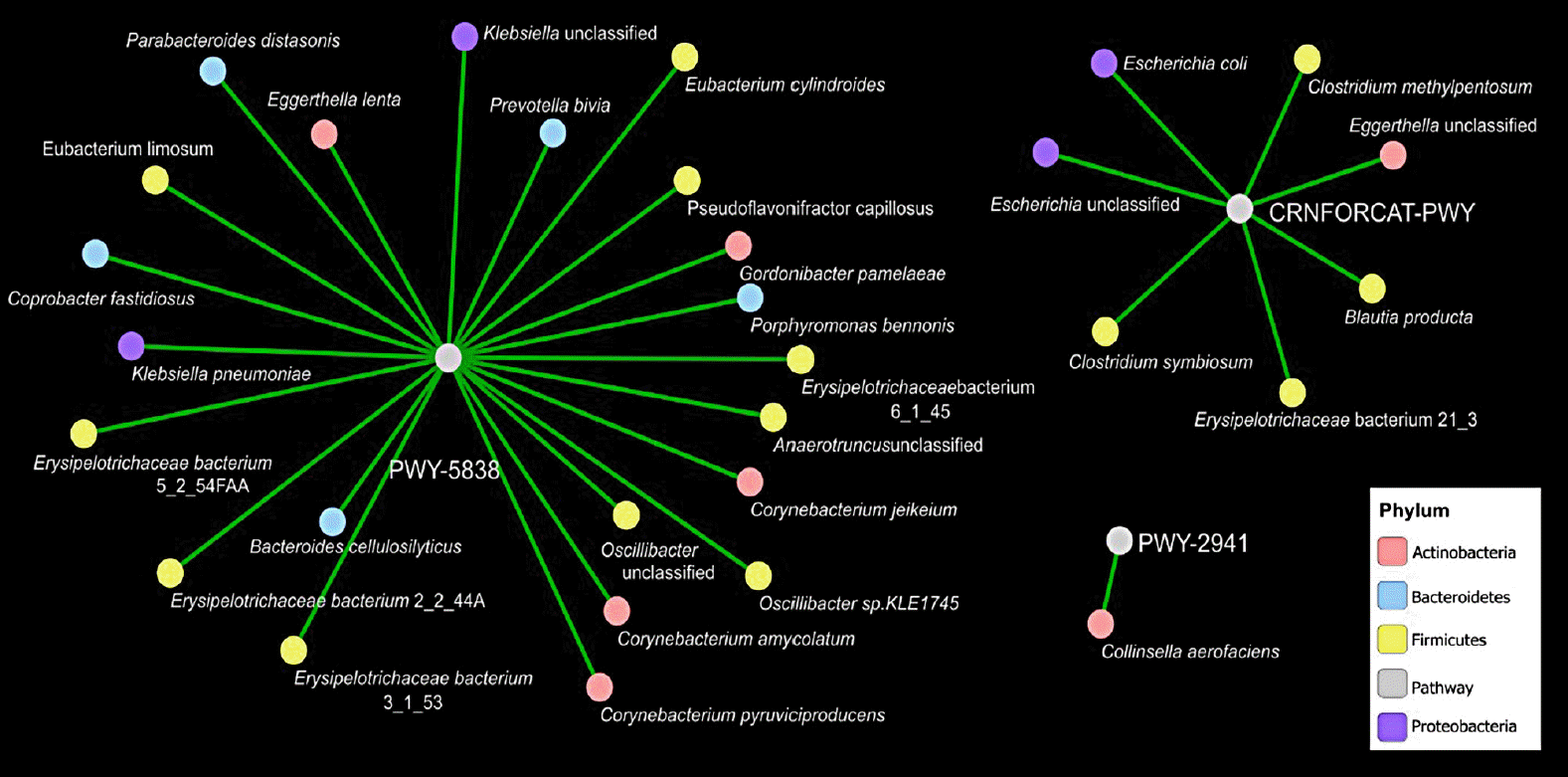
Fig. 4
Covariance network of bacterial genera and metabolic pathways. Panel a shows a force-driven network based on the predicted covariances (analysis of variance with P<0.05) between the genera and metabolic pathways identified as statistically altered in type 2 diabetes mellitus as compared to healthy.
![]()
DISCUSSION
In this study, we report a shift in gut microbiome function among individuals affected by T2DM as compared to healthy controls. Specific profiles of gut microbiome have been linked to T2DM across the globe based on variation in diet, probiotic use, and ethnicity among others [2627]. To overcome this issue, we normalized for fiber diet content, probiotic use, ethnicity and BMI in our study subjects to profile metabolic capabilities of microbiome corresponding to T2DM and healthy individuals. Interestingly, while exploring the community composition among microbiome, we did not detect significant differential abundance between the healthy and diabetic patients. These unexpected results perhaps attributed to small sample size in our study, despite high sequencing counts at an average of 10,164,901 quality-filtered reads per sample. Moreover, one of the potential confounders of our study population was the notable age difference between T2DM and healthy subjects (Supplementary Table 2), that may have an effect over microbiome composition and metabolic profiles that warrants future investigation [28]. Similarly, while we matched gender for our test group (T2DM), we were limited in our gender selection for the control group. We believe gender-specific differences in gut microbiome composition can also be a possible confounder [29]. Additionally, all T2DM individuals in our study population were under Dipeptidyl peptidase-4 inhibitors and metformin treatment that can modulate and affect their microbial diversity [30].
In contrast to what has previously been reported regarding decrease in the gut microbiome biodiversity with T2DM, we showed higher functional diversity (richness of unique pathways) among T2DM group. Our results suggest an important functional role for the altered microbiome to support their host against metabolic and inflammatory derangement of diabetes (Fig. 2). This gene enrichment in diabetic patients does not necessarily mean a healthy microbiome, but denotes a higher metabolic capability during low-grade inflammation [3132].
In this study, we characterized for the first time on a possible microbiome micronutrients mechanism to improve host insulin resistance and inflammation. We unveiled enrichment of super pathway of menaquinol-8 biosynthesis I (vitamin K2) (PWY-5838) at 30 log-2-fold differences when compared differential abundance between healthy and diabetic subjects, suggesting an important role of this pathway in insulin resistance and inflammation (Fig. 3).
Menaquinones (vitamin K2) are considered essential for humans and usually supplemented from nutrient sources and gut bacteria such as Escherichia coli, Bacteroides species, and some gram-positive bacteria [3334]. Previous observational and interventional studies have suggested a key role for vitamin K2 improving insulin resistance among other anti-inflammatory properties such as suppressing nuclear factor κB (NF-κB) signaling pathway [141535363738]. A prospective cohort study following up 38,094 individuals over 10 years concluded that menaquinones intake is associated with reduced risk of T2DM [14]. Others have suggested that vitamin K2 supplementation for few weeks affects β-cell function and/or insulin resistance in healthy subjects [1538]. That said, little is known about the direct link between gut microbiome composition and their contributions to menaquinones production in response to inflammatory conditions such as diabetes.
Interestingly, identification of highly immunogenic taxa Erysipelotrichaceae and Corynebacterium as a biomarker of T2DM, underscores that changes in relative abundance of certain taxa in the presence of a gut disorder may not reflect a taxa-specific functional role. Erysipelotrichaceae is a gram-positive bacterium that considered highly immunogenic and implicated in inflammatory bowel diseases [394041]. In addition, and consistent with our data shown in Supplementary Table 1, higher levels of Erysipelotrichaceae with obese individuals have been documented elsewhere [42]. Here, we report an important role of Erysipelotrichaceae and Corynebacterium in vitamin K2 production in T2DM subjects. These finding might be useful for new diabetes management strategies to improve insulin resistance.
In addition, we identified augmentation in L-lysine biosynthesis II (lysine acetylation) (PWY-2941) with high HbA1c T2DM group at approximately one log-2-fold differences when compared differential abundance between low and high HbA1c diabetic subjects, suggesting an important role the gut microbiome in preventing long-term complications of diabetes [43]. In contrast, creatinine degradation I was suppressed at approximately one log-2-fold differences among high HbA1c subjects when compared with low HbA1c diabetics, presumably as a result of altered gut microbiome failing to facilitate creatinine clearance in more complicated diabetic subjects (high HbA1c) [44].
In conclusion, one can hypothesize that manipulating the intestinal microbiome profiles of especially at-risk individuals might help them to avoid T2DM and other metabolic disorders. We hope that future functional studies will shed more light on microbiome micronutrient contribution to host health and disease conditions.
 XML Download
XML Download