

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Coffin–Siris syndrome (CSS; OMIM 135900) is a rare, clinically and genetically heterogeneous disorder [1]. Several genes encoding components of the BRG1/BRM-associated factor (BAF) chromatin remodeling complex are involved in CSS development [1]. AT-rich interaction domain 1A (ARID1A) is one of the largest core subunits of the BAF complex. ARID1A harbors an N-terminal DNA-binding ARID domain and a C-terminal folded region, recently annotated as the BAF250_C domain. Of the cases of molecularly-confirmed CSS reported worldwide, 5–7% have been attributed to pathogenic variants in ARID1A [2, 3]. Heterozygous loss-of-function (LoF) variants of ARID1A cause CSS [1-3]. Reported ARID1A variants are distributed throughout ARID1A, with no major site [1-7]. We present an extremely rare case of CSS accompanied by a de novo heterozygous missense variant cluster. The Institutional Review Board of Nowon Eulji Medical Center, Seoul, Korea, approved the use of human clinical materials and blood in this study (approval number: EMCS 2018-11-035). Written informed consent for the publication of medical images and genetic test results was obtained from the patient’s parents.
A six-year-old girl visited our clinic at Nowon Eulji Medical Center in August 2019, with a first generalized tonic seizure provoked by fever. She was the second child of non-consanguineous parents of Korean descent. She was born prematurely at 35 weeks and five days of gestation via vaginal delivery, with a low birth weight of 1,750 g (<3rd centile). Initial evaluation for multiple congenital abnormalities revealed a large perimembranous ventricular septal defect (VSD), patent foramen ovale (PFO), infantile hypertrophic pyloric stenosis (IHPS), left bifid ureter, double inferior vena cava, and uterine didelphys. She underwent pyloromyotomy for IHPS at the age of one month and VSD Dacron patch closure and PFO direct closure at the age of two months. She underwent surgical correction for bilateral congenital esotropia at 17 months and surgical removal of a left congenital cholesteatoma at 57 months. She has been receiving low-dose thyroid hormone (levothyroxine 25 μg/day) for subclinical hypothyroidism since four years of age.
From early infancy, she showed recognizable psychomotor developmental delay (DD) with hypotonia and hypermobile joints. She exhibited apparent language DDs, with receptive language being less affected than expressive language. She showed moderate intellectual disability (full-scale intelligence quotient, ~40) [8]. Physical examination at six yrs of age revealed normal growth (Fig. 1).
 | Fig. 1Photograph and magnetic resonance imaging and electroencephalography results of the patient at six years of age. (A, B) She has distinctive, coarse facial features, including a low frontal hairline, broad eyebrows, long eyelashes, puffy eyelids, a depressed nasal bridge, anteverted nares, a wide mouth, a thick and everted lower lip, and low-set ears with dysmorphic pinnae. (C, D) She has short distal phalanges of the fifth fingers and fifth toes and clinodactyly of the fifth fingers. (E, F) T1-weighted axial and T2-weighted sagittal brain magnetic resonance images show a nearly 6.6-cm arachnoid cyst in the posterior fossa (indicated by red arrows) and a short splenium of the corpus callosum (indicated by a blue arrow). (G) The electroencephalography images show interictal epileptiform discharges during sleep on right posterior head lesions.
|
For whole-exome sequencing, SureSelect Human All Exon V5 (Agilent Technologies, Santa Clara, CA, USA) was used for library preparation, and sequencing was performed on the NextSeq500 platform (Illumina Inc., San Diego, CA, USA) at GC Genome (Yongin, Korea). A cluster of ARID1A heterozygous missense variants, including c.5954C>G (p.Ser1985Cys), c.6314C>T (p.Thr2105Ile), c.6334C>T (p.Leu2112Phe), and c.6843G>T (p.Leu2281Phe), was identified based on the reference sequence NM_006015.6. The presence of these variants in the patient was confirmed by Sanger sequencing; neither parent had the variants (Fig. 2A). The biological parent-child relationship was confirmed using a short tandem repeat-based DNA test. Long-range PCR followed by TA cloning and Sanger sequencing were performed (Fig. 2B). Long-range PCR was performed using a primer set (forward, 5´-CCTGATGGACCTCCAGAAAA-3´; reverse, 5´-ACAGAAAGGCGTGAGGTGAT-3´) encompassing all variants in exon 20 of ARID1A, and the predicted amplicon size was 1,365 bp. All four variants were classified as likely pathogenic based on PS2 (de novo and paternity/maternity statuses confirmed), PM2 (absent from the population datasets dbSNP, gnomeAD, ExAC, 1000 Genomes, and KRGDB), and PP4 (highly specific phenotype) [9]. There were no possibly pathogenic variants in other genes, including those associated with CSS and other disorders with clinical features overlapping with those of CSS.
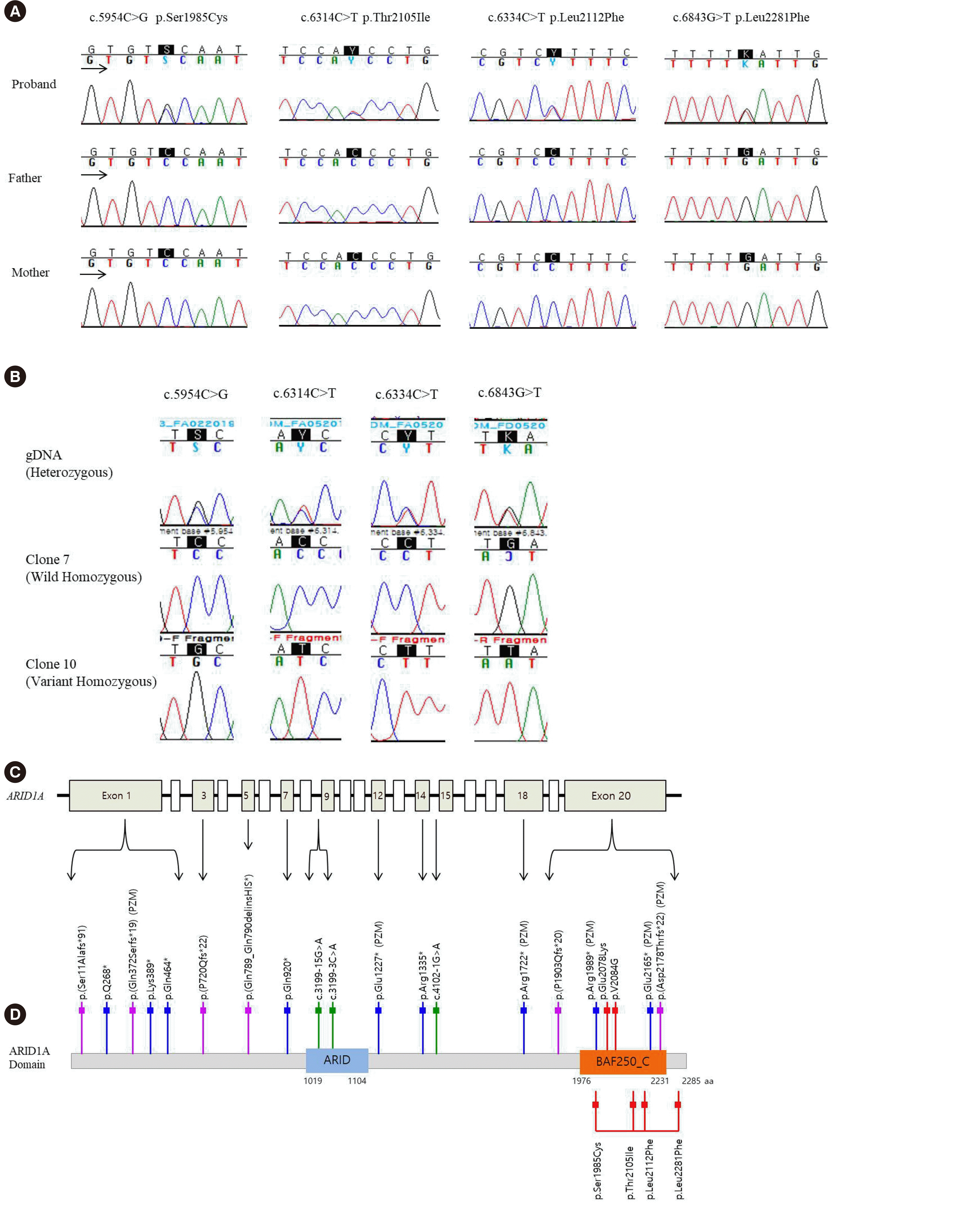 | Fig. 2Sanger sequencing and schematic view of the protein domain, all coding exons, and localization of all four novel ARID1A variants. (A) Sanger sequencing chromatograms of the four novel heterozygous variants c.[5954C>G;6314C>T;6334C>T;6843G>C] of ARID1A (NM_006015.6) in the patient and the wild-type genotype in her unaffected parents. (B) All four variants occurred in cis configuration. (C) ARID1A contains 20 exons and encodes the ARID1A protein, which contains 2,285 amino acids. (D) The ARID1A domain, according to UniProt (https://www.uniprot.org/uniprot/O14497) and Pfam (http://pfam.xfam.org/protein/O14497) databases showing previously reported ARID1A variants in CSS patients (upper panel) base on the HGMD Professional database (http://www.hgmd.cf.ac.uk/ac/all.php)and the four co-occurring variants identified in our patient (lower panel). To date, 18 heterozygous truncating ARID1A variants have been reported, including nine nonsense, six frameshift, and three splicing variants. Two missense variants have been indicated to be possibly associated with CSS. Purple bar indicates frameshift variant; blue bar indicates nonsense variant; green bar indicates splice-site variant; red bar indicates missense variant.
Abbreviations: ARID, AT-rich interaction domain; BAF250_C domain, C-terminal folded region; CSS, Coffin–Siris syndrome.
|
In autosomal dominant disorders, the co-occurrence of several variants of one causative gene is unusual and considered benign because of the high possibility of the variants occurring in trans configuration, even if they arose de novo [10]. Moreover, the homozygous loss of ARID1A is embryonic lethal in mice, suggesting that truncating germline ARID1A variants might be embryonic lethal in humans as well [3]. Here, four co-occurring variants of ARID1A arose de novo, which were ascertained to be in cis configuration in exon 20. We assumed that the missense variant cluster, located within 890 bp in cis configuration, caused an LoF effect in our patient.
Interestingly, three of the four ARID1A variants in our patient were within the BAF250_C domain (Fig. 2C, D) and were predicted to have deleterious effects by multiple means of computational evidence, including SIFT (http://sift.jcvi.org), PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2), and MutationTaster (http://www.mutationtaster.org).
This extremely rare case of classic CSS accompanied by a novel de novo heterozygous ARID1A missense variant cluster in cis c.[5954C>G;6314C>T;6334C>T;6843G>C] across the BAF250_C domain contributes to the elucidation of the genetic basis of CSS caused by ARID1A variants.
Go to : 
 XML Download
XML Download