

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Nasal polyps (NPs) are inflammatory outgrowths of the sinonasal epithelium, most commonly associated with chronic rhinosinusitis (CRS).1 Several hypotheses about the pathophysiology of NPs have been suggested and epithelial cell proliferation may be involved in the pathogenesis of NPs.2
Glucocorticoids, including dexamethasone, are the most widely prescribed anti-inflammatory drugs and are the only currently available treatment option with proven efficacy for NPs.3 According to previous reports, glucocorticoids increase apoptosis in various inflammatory cells including eosinophils, fibroblasts, and nasal epithelial cells.456
Estrogen (17β-estradiol) is known to regulate the expression of genes involved in cell survival, proliferation, differentiation, and reproduction,7 through binding to its receptors (ERs), ER-α, ER-β. The two ER isoforms have been detected in the cytoplasm and/or nuclei of glandular cells, epithelia, blood vessels, and inflammatory cells of the nasal mucosa.891011 Previous studies have shown that estrogen stimulates cell survival through nongenomic ER-α signaling, but induces cell death through nongenomic ER-β signaling.12 However, the role of ER-α in relation to cell survival in nasal cavity mucosa has not been clarified.
Therefore, in this study, we investigated the expression of ER-α in the inferior turbinate mucosa and NPs, and also investigated whether ER-α is related to cell survival using RPMI 2650 cells partially having the characteristics of the human nasal mucosa. In addition, if ER-α is related to cell survival, we tried to determine how dexamethasone affects the expression of ER-α and apoptosis when dexamethasone is treated to RPMI 2650 cells.
The RPMI 2650 cells used in this study have been shown to closely resemble normal human nasal epithelium with respect to their karyotype, several cytokeratin expression patterns, and the presence of mucoid material on the cell surface.1314
Go to : 
METHODS
Patients
Based on the definition in a 2007 European position paper on rhinosinusitis and NPs,9 we recruited patients with a diagnosis of CRS with NPs undergoing elective endoscopic sinus surgery at our hospital from September 2018 to January 2019. Patients with malignancies, fungal sinusitis, and atopy were excluded from the study. The recruited patients did not use intranasal steroid spray for at least four weeks before surgery. NP tissues (n = 15) for Western blots and immunohistochemistry were obtained from endoscopic sinus surgery. Normal nasal mucosae (n = 15) for Western blots and immunohistochemistry were isolated from the healthy inferior turbinates of patients who had no allergic rhinitis and underwent septal surgery or rhinoplasty.
Immunohistochemical staining
Stored formalin-fixed and paraffin-embedded samples taken from NPs and inferior turbinate tissues were used for immunohistochemical staining. To perform immunohistochemical staining for ER-α, a 4-μm-thick paraffin block was deparaffinized and cleared in xylene, rehydrated with ethanol, and incubated in citrate buffer (0.01 M, pH 6.0), with subsequent heating using an microwave for 15 minutes. The sections were incubated overnight at 4°C with anti-ERα antibody (sc-8002; Santa Cruz Biotechnology, Dallas, TX, USA), followed by incubation in biotin-labeled secondary antibody for 30 minutes and then streptavidin-peroxidase for another 30 minutes. The staining was visualized using 3,3′-diaminobenzidine tetrahydrochloride.
Cell culture
The RPMI 2650 cells (American Type Culture Collection, Manassas, VA, USA) were maintained in Eagle's Minimum Essential Medium (EMEM) supplemented with 5% fetal bovine serum, 1 mM glutamine, 100 units of penicillin/mL, and 100 μg of streptomycin/mL. The cells were grown to 70% confluence in a monolayer culture under submerged conditions for 24 hours before treatment. They were harvested by trypsinization for Western blot analysis, the MTT assay, and the annexin V-phycoerythrin (PE) binding assay.
Western blot analysis
A portion of the specimens was frozen in liquid nitrogen immediately after surgical resection and stored at −80°C for Western blotting. The tissue samples or RPMI 2650 cells were homogenized in RIPA buffer (1× phosphate-buffered saline, 1% NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 10 μg/mL phenylmethanesulfonyl fluoride, and a protease inhibitor cocktail tablet). Protein concentrations were determined using a Pierce BCA Protein Assay kit (Thermo Scientific, Rockford, IL, USA), according to the manufacturer's protocol. Cell and tissue lysates containing 40 μg of protein were resolved on NuPAGE 4%–12% bis-tris polyacrylamide gels and then electrotransferred onto a polyvinylidene difluoride membrane. The membrane was probed with 1:1,000 diluted primary antibodies in casein blocking buffer at 4ºC for 16 hours, followed by incubation for 2 hours at room temperature with a 1:5,000 dilution of secondary antibody coupled to horseradish peroxidase in casein blocking buffer. The signals were detected using an enhanced chemiluminescence detection kit (Invitrogen, Carlsbad, CA, USA). The membranes were stripped and reprobed with anti-β-actin antibody (A2228; Sigma-Aldrich, St. Louis, MO, USA) as a loading control. The antibodies used were specific antibodies against ER-α (sc-543; Santa Cruz Biotechnology), Mcl-1 (4572; Cell Signaling Technology, Danvers, MA, USA), Bcl-2 (2820; Cell Signaling Technology), BAX (5023; Cell Signaling Technology), PARP (9542; Cell Signaling Technology), procaspase-3 (9665; Cell Signaling Technology), and cleaved caspase-3 (9664; Cell Signaling Technology). The secondary antibodies used were HRP-conjugated anti-rabbit IgG (sc-2004; Santa Cruz Biotechnology), anti-goat IgG (sc-2020; Santa Cruz Biotechnology), and anti-mouse IgG (sc-2005; Santa Cruz Biotechnology).
MTT assay
For the cell viability assay, 96-well microtiter plates were seeded with approximately 5,000 cells per well and incubated with dexamethasone (D4902; Sigma-Aldrich) and ICI 182780 (I4409; Sigma-Aldrich), a selective ER-α antagonist, alone or in combination, in EMEM for the indicated times, followed by exposure to tetrazolium dye (final concentration = 0.1 mg/mL) for 4 hours at 37°C. The absorbance at 540 nm was measured using a GloMax-Multi Microplate Multimode Reader (Promega, Madison, WI, USA). The number of viable cells was expressed as a percentage by comparing the results with those of the vehicle-treated control cells (100%) for each treatment and time point.
Annexin V-PE binding assay
The distribution of apoptotic cells was determined with a Muse Annexin V and Dead Cell Assay Kit (Merck KGaA, Darmstadt, Germany), according to the manufacturer's protocol. This kit included a fluorescent dye (PE) conjugated to annexin V to detect phosphatidylserine on the external membrane of apoptotic cells and 7-amino-actinomycin D as a dead cell marker. Briefly, the cells (1 × 105 cells/well) were seeded into a 6-well culture plate and incubated with dexamethasone and ICI 182780, alone or in combination, in EMEM for 48 hours at 37°C. The cells were trypsinized and collected in culture medium, mixed with the MuseTM Annexin V and Dead Cell reagent (Merck KGaA), and analyzed using a Muse Cell Analyzer (Merck KGaA).
Statistical analysis
All values are expressed as the mean ± standard deviation (SD). The Mann-Whitney U 2-tailed test was used for between-group comparisons. Differences in the proportions between the groups were analyzed by the χ2 test or Fisher's exact test, as appropriate. A difference of P < 0.05 was considered statistically significant.
Ethics statement
Informed consent was obtained from each patient and the study was approved by the Institutional Review Board of Cheonan Hospital of Soonchunhyang University (approval No. 2018-05-061).
Go to :
RESULTS
The NP group was comprised of 15 patients (eight males and seven females) with a mean age (± SD) of 41.4 ± 14.8 years. The control group consisted of 15 healthy individuals (seven males and eight females) with a mean age (± SD) of 39.8 ± 13.4 years. There was no significant difference between the mean age and sex of the participants in the two groups.
Immunohistochemistry
Immunohistochemical staining showed the presence of ER-α within many cells of the NPs and healthy inferior turbinate mucosae (Fig. 1). ER-α immunoreactivity in the NP specimens was observed in the cytoplasm and/or nuclei of the epithelial cells, submucosal glands, blood vessels, and inflammatory leukocytes (Fig. 1A-C). The expression pattern of ER-α in the inferior turbinate mucosa was similar to that in the NPs, but the immunoreactivity was weaker than that in the NPs (Fig. 1D).
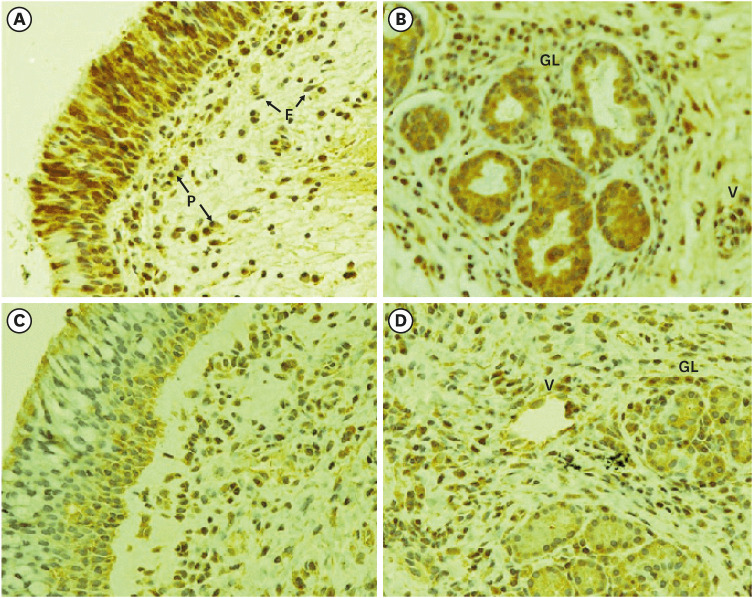 | Fig. 1Immunohistochemical staining of ER-α expression in nasal polyps (A, B) and healthy inferior turbinate mucosa (C, D). Immunohistochemical reactions for ER-α in the nuclei and cytoplasm of the respiratory epithelium, submucous glands, venules, and inflammatory cells of the lamina propria (original magnification: 400×).ER = estrogen receptor, P = plasma cells, F = fibroblast, GL = submucous gland, V = venule.
|
Western blot analysis
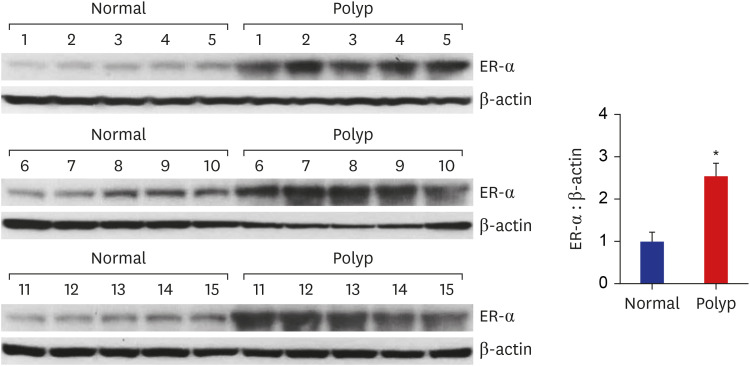
To validate the results of the immunohistochemical staining, ER-α protein expression was examined by Western blot analysis in the NPs and healthy inferior turbinate mucosae obtained from 30 patients who had undergone surgery. As shown in Fig. 2, an ER-α protein band was detected in all tissues examined. The upregulation of ER-α protein expression was observed in 13 out of 15 NP tissues compared to the healthy inferior turbinate mucosae.
Relationship between ER-α and cell survival in RPMI 2650 cells
To determine whether ER-α is related to cell survival in RPMI 2650 cells, the cells were exposed to ICI 182780 for 24, 48, and 72 hours and examined using the MTT assay. As shown in Fig. 3, treatment with ICI 182780 inhibited the growth of RPMI 2650 cells in concentration- and time-dependent manners.
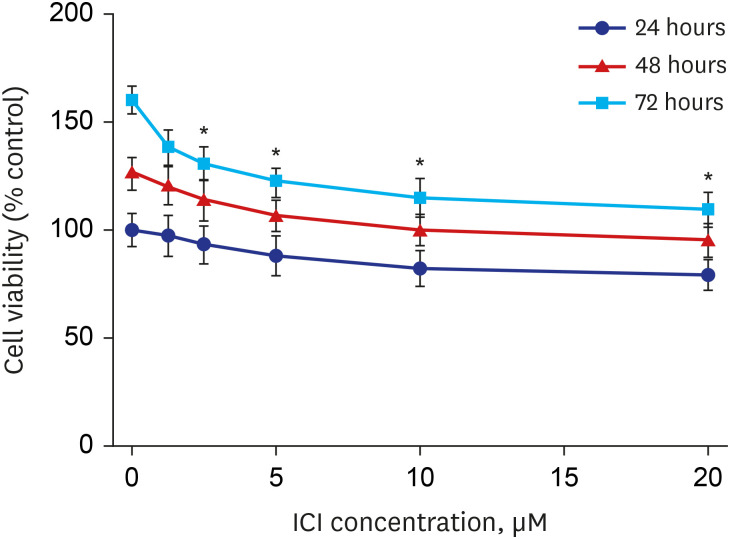 | Fig. 3The effect of ICI 182780 on RPMI 2650 cell viability. The cells were treated with increasing concentrations of ICI 182780 (0, 1.25, 2.5, 5, 10, and 20 μM) for 24, 48, and 72 hours. The percentage of viable cells was then determined using the MTT assay. The error bars represent the mean ± standard deviation of three independent experiments.ICI = ICI 182780.
*P < 0.05 vs. untreated controls.
|
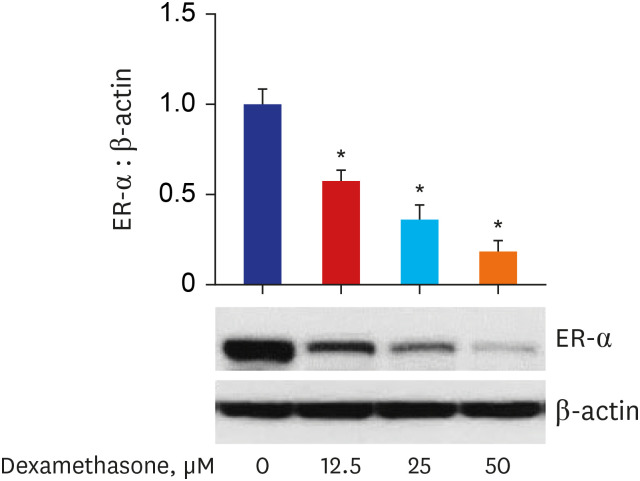
Effect of dexamethasone on ER-α expression in RPMI 2650 cells
After the RPMI 2650 cells were treated with dexamethasone, the expression of ER-α was measured. As shown in Fig. 4, Western blot analysis showed that dexamethasone treatment decreased the levels of ER-α protein in a concentration-dependent manner.
Effect of dexamethasone on cell survival in RPMI 2650 cells
The sensitivity of RPMI 2650 cells to dexamethasone was examined using the MTT assay after exposure to dexamethasone for 24, 48, and 72 hours. As shown in Fig. 5, treatment with dexamethasone inhibited the growth of RPMI 2650 cells in a concentration- and time-dependent manner.
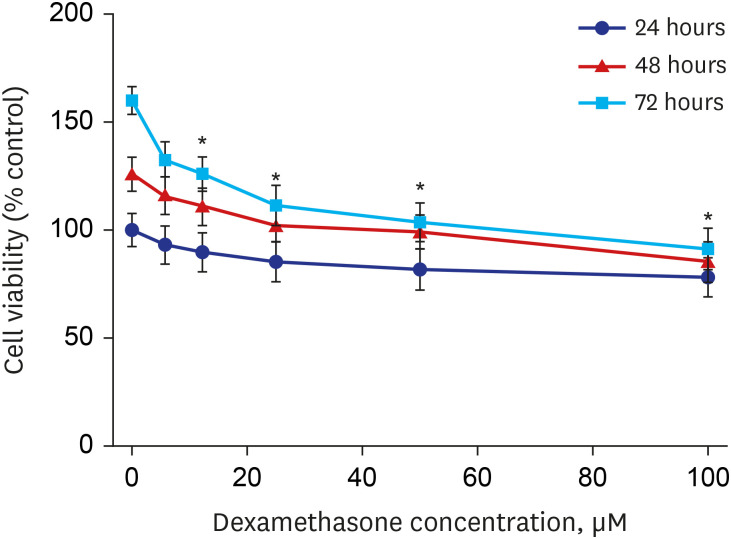 | Fig. 5The effect of dexamethasone on RPMI 2650 cell viability. The cells were treated with increasing concentrations of dexamethasone (0, 6, 12.5, 25, 50, and 100 μM) for 24, 48, and 72 hours. The percentage of viable cells was then determined using the MTT assay. The error bars represent the mean ± standard deviation of three independent experiments.
*P < 0.05 vs. untreated controls.
|
Next, doses of 6, 12.5, and 25 µM dexamethasone and 2.5, 5 µM ICI 182780 were chosen for combination treatments. The combined treatments of dexamethasone and ICI 182780 resulted in a more significant decrease in the viability (approximately 58.3%) of RPMI 2650 cells compared to 5 µM ICI 182780 (approximately 23.6%) or 25 µM dexamethasone (approximately 30.3%) treatment alone (Fig. 6).
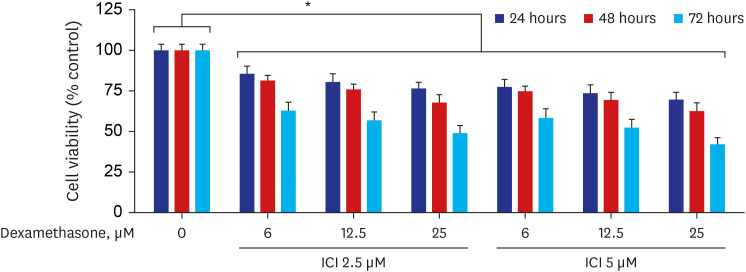 | Fig. 6The effects of combined treatment with dexamethasone and ICI 182780 on RPMI 2650 cell viability. The cells were treated with combinations of ICI 182780 (2.5 and 5 μM) and increasing concentrations (0, 6, 12.5, and 25 μM) of dexamethasone for the indicated times. The percentage of viable cells was then determined using the MTT assay. The error bars represent the mean ± standard deviation of three independent experiments. ICI, ICI 182780.
*P < 0.05 vs. the respective untreated controls.
|
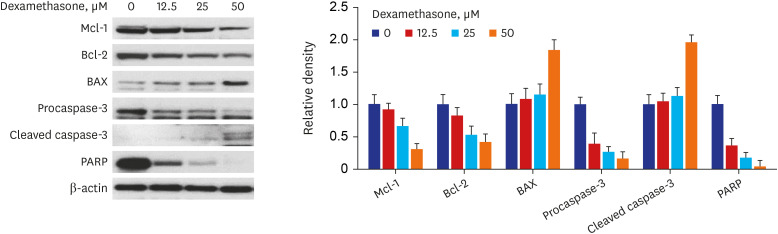
Effect of dexamethasone on apoptosis in RPMI 2650 cells
Apoptosis-related proteins were measured in RPMI 2650 cells treated with dexamethasone. As shown in Fig. 7, Western blot analysis showed that anti-apoptotic proteins, including Bcl-2 and Mcl-1, were downregulated, whereas the pro-apoptotic protein BAX was upregulated. In addition, the levels of active (cleaved) caspase-3 as an apoptotic marker were increased and those of its downstream substrate, PARP, were decreased.
To further elucidate whether the growth inhibition of RPMI 2650 cells by dexamethasone was related to apoptotic cell death, apoptosis was analyzed in dexamethasone-treated RPMI 2650 cells using annexin V-PE staining. As shown in Fig. 8, the proportion of annexin V-PE positive cells undergoing apoptosis increased as the dexamethasone concentration increased. Moreover, the annexin V-PE positive cells increased to approximately 35.45% after combined treatment with dexamethasone and ICI 182780 compared to 25 µM dexamethasone (approximately 17.13%) or 5 µM ICI 182780 (approximately 15.82%) alone.
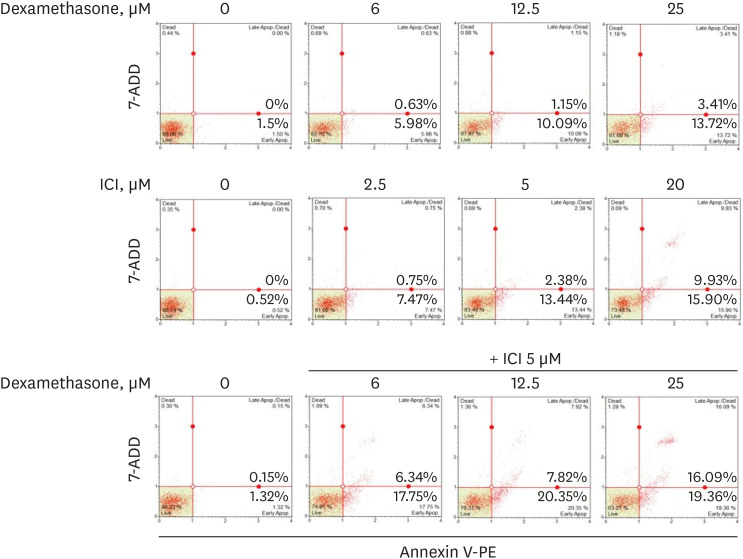 | Fig. 8The effects of dexamethasone and ICI 182780 treatment on RPMI 2650 cell death. The cells were treated with increasing concentrations of dexamethasone (0, 6, 12.5, and 25 μM) and ICI 182780 (0, 2.5, 5, and 20 μM), alone or in combination, for 48 hours. The percentage of apoptotic cells after annexin V-phycoerythrin binding was determined using a Muse cell analyzer.ICI = ICI 182780, PE = phycoerythrin.
|
Go to :
DISCUSSION
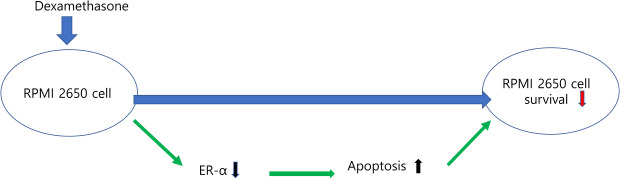
In this study we found that ER-α was more highly expressed in NPs than in healthy inferior turbinate mucosae and ER-α was associated with cell survival in RPMI 2650 cells. When RPMI 2650 cells were treated with dexamethasone, ER-α expression was downregulated and apoptosis of RPMI 2650 cells were increased. Decreased ER-α expression likely contributed to the induction of apoptotic cell death in the RPMI 2650 cells.
Some studies have characterized the presence of ER-α and ER-β in several organs not associated with reproduction.810151617 According to previous reports,81015 ER-α is mainly expressed in the cytoplasm of cells of the seromucinous glands, venules, and/or the nuclei of interstitial cells (mainly mast cells) in the nasal mucosa. In our study, ER-α immunoreactivity was mainly found in the cytoplasm and/or nuclei of respiratory epithelia, submucosal glands, and inflammatory cells, including fibroblasts. The difference in these results may be due to differences in the analytical methods used.
In the present study, the immunoreactivity of ER-α was stronger and more highly expressed in NPs than in normal inferior turbinate mucosae. This finding was confirmed by Western blot analysis. ER-α protein expression was significantly upregulated in 13 of 15 NP tissues compared to the expression levels in normal inferior turbinate mucosae. These results are the first to objectively show that ER-α expression was significantly elevated in NPs compared to normal inferior turbinate mucosae, suggesting that ER-α plays a specific role in the development of NPs. Meanwhile, in this experiment, NPs were not separately classified according to endotype. Therefore, it should be kept in mind that the expression pattern of ER-α may differ depending on the subtypes of NP.
To determine the role of ER-α in the nasal mucosa, we investigated the relationship between ER-α expression and cell survival using the ER inhibitor ICI 182780 in RPMI 2650 cells. The results showed that ER-α was associated with cell survival. Therefore, we speculate that estrogen stimulates epithelial cell survival through ER-α signaling in the nasal cavity mucosa.
Nasal epithelial cells not only act as a structural barrier but also play an important role as a potent source of inflammatory mediators, including various cytokines, chemokines, and cell adhesion molecules.18
Glucocorticoids, such as dexamethasone, are drugs used for the first-line treatment of CRS with NPs to suppress nasal inflammation.3 The clinical efficacy of glucocorticoids for treating NPs results from the induction of apoptosis in both eosinophils and T-lymphocytes that infiltrate NPs and the downregulation of epithelial GM-CSF production, which prolongs eosinophil survival.4
Dorscheid et al.19 described the dexamethasone-induced apoptosis of bronchial epithelial cells in both primary and immortalized cell lines. In contrast, corticosteroids were reported to protect mammary gland epithelial cells against apoptotic stimuli20 and inhibit apoptosis induced by Fas ligand in alveolar epithelial cells.21 Thus, the effects of dexamethasone on apoptosis were demonstrated to vary depending on the target tissue.
There has been minimal research documenting the effects of corticosteroids on cell survival or apoptosis in nasal epithelial cells.622 One study showed that treating human primary nasal epithelial cells with dexamethasone induced apoptosis in a concentration-dependent manner.6 However, the report did not mention how dexamethasone induces apoptosis in nasal epithelial cells.
In our study, we found that dexamethasone inhibited the growth of RPMI 2650 cells in concentration- and time-dependent manners. Furthermore, when RPMI 2650 cells were treated with dexamethasone, pro-apoptotic proteins BAX and cleaved caspase-3 were upregulated and anti-apoptotic proteins Bcl-2 and Mcl-1 were downregulated as the concentration of dexamethasone increased. These results suggest that the decreased cell viability in dexamethasone-treated RPMI 2650 cells may be associated with increased apoptotic cell death. This inference was confirmed by the results of experiments in which the percentage of apoptotic cells was increased as the concentration of dexamethasone increased. Thus, these findings indicate that dexamethasone had a pro-apoptotic effect in RPMI 2650 cells. This observation is consistent with a study by Bobic et al.,6 showing that dexamethasone induced apoptosis in primary nasal epithelial cells.
To date, there have been some reports that dexamethasone reduces the expression of ER-α,232425 but there has been no study on the effect of dexamethasone on ER-α expression in airway epithelial cells. In this study, we found that the dexamethasone treatment of RPMI 2650 cells decreased the expression of ER-α as the concentration of dexamethasone increased.
We also found that ER-α was associated with the survival of RPMI 2650 cells in this study. In addition, the expression of ER-α may be related to apoptosis in RPMI 2650 cells as shown by the increased percentage of apoptotic cells following treatment with the ER inhibitor ICI 182780.
The above results indicate that dexamethasone treatment of RPMI 2650 cells decreased cell viability, at least in part, through downregulating ER-α protein expression, possibly contributing to the induction of RPMI 2650 cell death.
Studies have suggested that epithelial cell proliferation may be involved in the pathogenesis of NPs.226 In the present study, dexamethasone treatment increased the apoptosis of RPMI 2650 cells, suggesting that corticosteroids, including dexamethasone, may play a role in reducing epithelial cell proliferation. In this regard, corticosteroids, which have been successfully used to prevent the recurrence of NPs after surgery, may act not only via anti-inflammatory pathways but also via the direct inhibition of epithelial cell proliferation.
Nasal mucosa or NPs are composed of various cells such as epithelial cells, glandular cells, vascular cells, and inflammatory cells. Therefore, it should be borne in mind that effects of corticosteroids at the tissue level such as NPs may differ from those at the cellular level such as RPMI 2650 cells.
This study had several limitations. First, in this study, we used an RPMI 2650 cell line cultured by submerging the cells in media rather than using primary nasal epithelial cells to analyze the response to dexamethasone. Although the RPMI 2650 cell line exhibits advantages, including good genetic stability, easy culturing, and good reproducibility of results,27 it is less morphologically and physiologically similar to normal nasal epithelium than primary nasal epithelial cells. Therefore, in this experiment, if primary nasal epithelial cells cultured by an air-liquid interface method were used instead of the RPMI 2650 cells, the results could better reflect actual nasal mucosa. Second, we used an antagonist of ER-α to investigate the effects of ER-α on cell survival in RPMI 2650 cells and found that cell viability was reduced. However, to accurately determine the effect of ER-α on cell survival, a process that demonstrates cell viability increases when an ER-α agonist is used is required.
In conclusion, we demonstrated that the expression of ER-α was increased in NPs compared to healthy inferior turbinate mucosa. ER-α expression levels were related to cell survival in RPMI 2650 cells. It is not known by which pathway apoptosis was increased when dexamethasone was treated to RPMI 2650 cells, but it is thought that a pathway that increases apoptosis through decreased ER- α expression may be included. Further studies are needed on the mechanism by which dexamethasone decreases the expression of ER-α in RPMI 2650 cells and conditions in which decreased ER-α expression leads to increased apoptotic cell death.
Go to :
 XML Download
XML Download