

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hearing loss (HL) in children, even when it is slight to mild,1 affects speech-language, social-behavior, cognitive, and emotional development, as well as attention and academic achievement.23 Even unilateral HL may impair school performance and lead to educational failure and deleterious behavior outcomes.4 Known to affect more than 5% of the general population,5 HL can be grouped into sensorineural hearing loss (SNHL), conductive hearing loss (CHL), and mixed form. SNHL is caused by abnormalities in the inner ear, cochlea, or the auditory nerve, and CHL results from the middle or outer ear diseases, commonly acute otitis media or otitis media with effusion (OME).67
HL is more prevalent in patients with chronic kidney disease (CKD) than in the general population.58 While the overall prevalence of HL with CKD is 40%–75% in adults and about 40% in children,910 SNHL is more common than CHL in this population,811 with the prevalence of SNHL in adult patients estimated to be 20%–87% and 29%–40% in children.1012 Several potential hypotheses have been proposed to explain the higher prevalence of HL in patients with CKD; one of them is that the kidney and the stria vascularis of the cochlea share physiologic, ultrastructural, and antigenic characteristics that could underlie the link between CKD and HL.810 In fact, several genetic diseases or syndromic diseases manifest both CKD and SNHL, such as Alport syndrome or branchio-oto-renal (BOR) syndrome,1314 as well as both CKD and CHL, such as BOR syndrome, Fraser syndrome, and CHARGE syndrome.151617 While there have been several studies on SNHL in children with CKD,1018 studies on CHL in children with CKD are hard to find.
On the other hand, prematurity is also a significant risk factor for HL in children.19 The prevalence of SNHL is up to 17.5% in very preterm newborns, which is much higher than that in late preterm or term newborns.1920 In addition to prematurity itself, other perinatal problems (PP) contribute to the risk of developing HL in preterm newborns, such as small for gestational age (GA), perinatal asphyxia, exposure to ototoxic drugs, and neonatal sepsis.1920 While prematurity is rather common in the pediatric CKD population,2122 the significance of prematurity in HL of pediatric CKD is yet to be known.
In this study, we assessed the prevalence and risk factors of HL, both SNHL and CHL, according to the underlying causes of childhood-onset CKD.
METHODS
Patients
This retrospective study was conducted at Seoul National University Children's Hospital, a tertiary referral teaching hospital in Korea. Patients with childhood-onset CKD (onset age of renal disease < 18 years), who were diagnosed as showing CKD stage 2–5 from 1990 to July 2019, were recruited via electronic medical record search. The clinical characteristics and history of the patients were reviewed, along with the renal function at HL detection and the last follow-up. The estimated glomerular filtration rate (eGFR) was calculated using cystatin C-based Schwartz equation23 for patients under 18 years of age, and the Modification of Diet in Renal Disease formula for patients aged 18 and over, and the CKD stage was determined using the Kidney Disease Outcomes Quality Initiative CKD classification.24 The patients were grouped according to the causes of CKD: congenital anomalies of the kidney and urinary tract (CAKUT), glomerulopathies (GP), cystic kidney diseases (CYST), perinatal problems (PP), and others. CYST included autosomal dominant and recessive polycystic kidney diseases and nephronophthisis (NPHP). PP included preterm birth, perinatal asphyxia, congenital anomalies of other organs including brain, heart, eye, maternal or congenital infections, sepsis, and other perinatal/neonatal problems, excluding those with other causes of CKD. According to the GA, patients were grouped into very preterm (GA < 32+0 weeks), moderately preterm (GA 32+0–33+6 weeks), late preterm (GA 34+0–36+6 weeks), and term (GA ≥ 37+0 weeks) babies.19 The use of diuretics and the history of seizure were not reviewed.
Hearing tests were performed in newborns (for congenital HL) and those with genetic diseases known to accompany HL as screening, and in patients with auditory symptoms such as middle ear diseases, aural fullness, or hearing difficulty. Audiometric examinations were performed by trained audiologists using pure tone audiometry (PTA) or the automated brainstem response test (ABRT) in young children who did not cooperate. Assessment of the hearing threshold was performed on the basis of bone-conducted (BC) signals and air-conducted (AC) signals. Hearing threshold was measured over frequencies of 0.5, 1, 2, and 4 kHz for both ears separately.7 The criteria for HL was based on the World Health Organization classification, defined as a hearing threshold greater than 25 decibel (dB) on PTA or ABRT results.25 SNHL was defined as an increase in both air and bone thresholds, while CHL was diagnosed when there was a difference of at least 15 dB between AC and BC thresholds at the same frequency. OME and acute otitis media were evaluated by otolaryngologists using tympanometry.
Syndromic HL was previously defined as a form of HL that presents with anomalies of systems other than the auditory system by Koffler et al.26 Similarly, HL that presented with anomalies of systems other than the renal and the auditory systems, or HL in CKD with a known syndrome or genetic mutation was referred to as “renal” syndromic HL in this study.
Statistical analyses
Statistical analysis was performed using the SPSS software version 20.0 (SPSS Inc., Chicago, IL, USA) and R statistics (R Foundation, Vienna, Austria). Data were presented as median (interquartile range [IQR]) or frequency (percent). Student t-test and paired t-test were used for the analysis of metric variables that met the criteria for normal distributions. The χ2 test and Mann-Whitney U-test were used for the analysis of metric variables and continuous variables, respectively. The linear-by-linear association test was used to determine the relationship between HL and CKD stages. Using R statistics, univariate and multivariate analyses were performed to analyze the risk factors of HL. In the multivariate logistic regression model, variables that showed statistical significance in univariate analysis were used, and a final model was derived using the backward selection method. A P value of < 0.05 was considered significant. In the univariate and multivariate analyses, the risk of HL was presented with odds ratio (OR) and 95% confidence interval (CI).
RESULTS
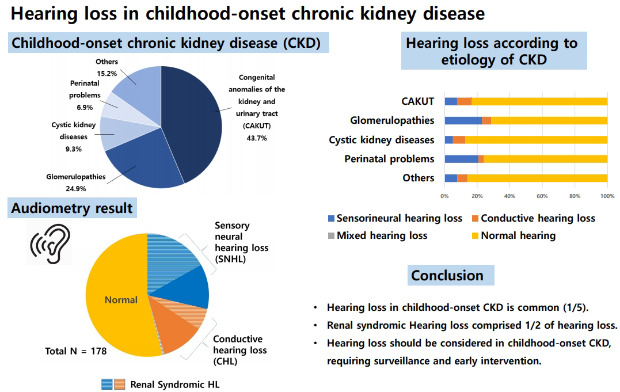
A total of 421 patients (male:female, 279:142; median age, 15.8 years [IQR, 10.9–19.2 years]) were reviewed. The median onset age of renal symptoms was 1.7 years (IQR, 0.0–7.3 years), and the median duration of follow-up was 8.4 years (IQR, 4.1–14.0 years). At the last follow-up, more than half of the patients (n = 241) had end-stage renal disease (ESRD), and their median age at ESRD was 9.0 years (IQR, 4.7–13.3 years). The underlying diseases for CKD in our population were CAKUT (n = 184, 43.7%), GP (n = 105, 24.9%), CYST (n = 39, 9.3%), PP (n = 29, 6.9%), and others (n = 64, 15.2%) (Table 1).
Table 1
Baseline characteristics of the patients (n = 421)
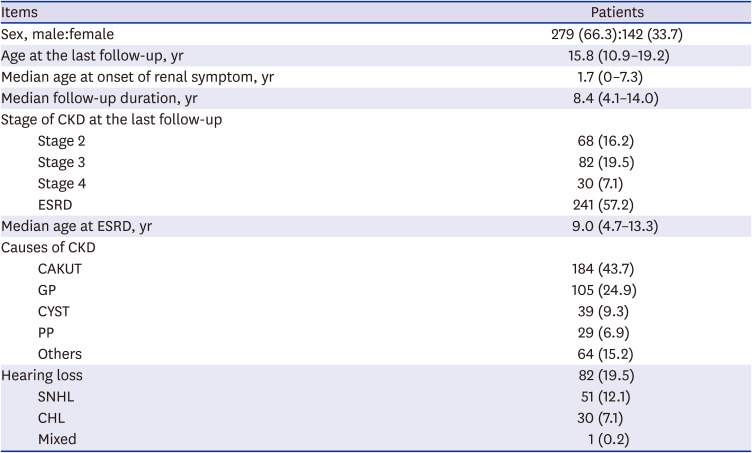
Values are presented as number (%) or median (interquartile range).
CKD = chronic kidney disease, ESRD = end-stage renal disease, CAKUT = congenital anomalies of the kidney and urinary tract, GP = glomerulopathies, CYST = cystic kidney diseases, PP = perinatal problems, SNHL = sensorineural hearing loss, CHL = conductive hearing loss.
Audiometric examinations were performed in 178 (42.3%) patients at a median age of 7.5 years (IQR, 2.25–11.23 years), and HL was detected in 82 (19.5%) patients, including SNHL in 51 patients (12.1%, bilateral in 40 patients), CHL in 30 patients (7.1%, bilateral in 23 patients), and mixed HL in 1 patient (0.2%, bilateral). HL was detected at a median age of 7.7 years (IQR, 4.5–12.1 years), and a median eGFR at detection of HL was 35.6 mL/min/1.73m2 (IQR, 9.6–65.1 mL/min/1.73m2). HL was more common as the CKD stage increased (P = 0.002), and CHL was more prevalent in ESRD than non-ESRD (P = 0.003) (Table 2). There was no difference in the prevalence of HL according to the GA (Table 2).
Table 2
Hearing loss according to CKD stage
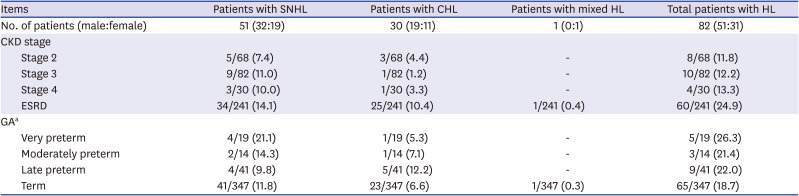
Values are presented as number (%).
SNHL = sensorineural hearing loss, CHL = conductive hearing loss, HL = hearing loss, CKD = chronic kidney disease, ESRD = end-stage renal disease, GA = gestational age.
aVery preterm (GA < 32+0 weeks), moderately preterm (GA 32+0–33+6 weeks), late preterm (GA 34+0–36+6 weeks), term (GA ≥ 37+0 weeks).
The prevalence of HL in each group was as follows: 16.8% (SNHL, 7.6%; CHL, 8.7%; mixed, 0.5%) in the CAKUT group, 28.6% (SNHL, 22.9%; CHL, 5.7%) in the GP group, 12.8% (SNHL, 5.1%; CHL, 7.7%) in the CYST group, 24.1% (SNHL, 20.7%; CHL, 3.4%) in the PP group, and 14.1% (SNHL, 7.8%; CHL, 6.3%) in the others group. The prevalence of HL and SNHL were higher in the GP (P = 0.007 and 0.021, respectively) than in the other groups (Fig. 1).
Fig. 1
Prevalence of hearing loss according to disease group. Numbers inside the columns indicate the numbers of patient. Proportion of renal syndromic HL is indicated by hatching.
CAKUT = congenital anomalies of the kidney and urinary tract, SNHL = sensorineural hearing loss, CHL = conductive hearing loss, HL = hearing loss.
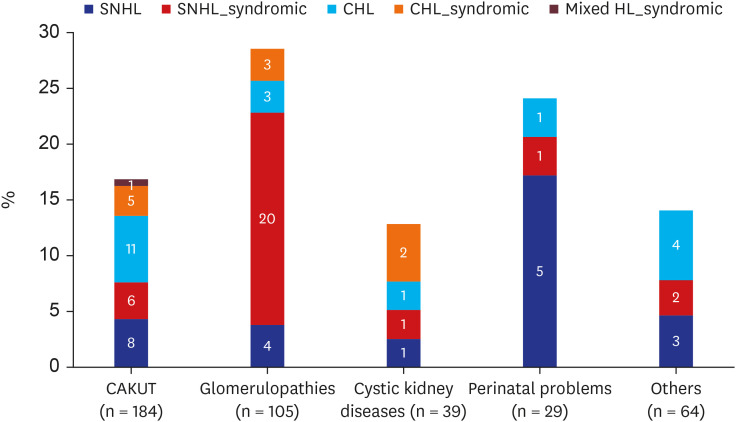
Of the 82 patients with HL, 41 (50.0%; SNHL:CHL:mixed HL, 30:10:1) had genetic or syndromic diseases, which was annotated as renal syndromic HL; these cases corresponded to 58.8% (30/51) of the cases of SNHL, 33.3% (10/30) of the cases of CHL, and 100% (1/1) of the cases of mixed HL (Fig. 1). Non-syndromic SNHL was caused by nephrotoxic agents (n = 5), PP (n = 5), and unknown causes (n = 11). Among CHL patients, 27/30 (90.0%) had OME. On follow-up audiometry, two-thirds patients (20/30) with CHL still had HL, and seven (23.3%) recovered. Follow-up audiometry was not available in the other three.
HL according to the underlying disease
CAKUT group
Among the 184 patients of the CAKUT group, 31 had HL (SNHL:CHL:mixed HL, 14:16:1). Among the 14 patients with SNHL, 4 patients had syndromes known to cause SNHL: BOR syndrome (n = 2), Townes-Brocks syndrome (n = 1), and Goldenhar syndrome (n = 1). A patient with renal tubular dysgenesis from ACE mutations and another patient with multiple congenital anomalies (no genetic diagnosis obtained) also had SNHL. Among the remaining 8 patients with SNHL, two cases could be attributed to ototoxic antibiotics, and we could not find any causes of SNHL in the remaining 6 patients. Fourteen of the 16 patients with CHL had OME, including 2 patients with the ossicular anomaly, 1 patient with CHARGE syndrome, 1 patient with Sotos syndrome, 1 patient with cholesteatoma and Sifrim-Hitz-Weiss syndrome, and another patient with multiple congenital anomalies. One of the two patients without OME had cholesteatoma and ossicular anomaly, and the other patient had Fraser syndrome. The patient with bilateral mixed HL was clinically diagnosed as having Baller-Gerold syndrome. Overall, 12 of 31 (38.7%; 6 SNHL, 5 CHL, and 1 mixed HL) patients with HL in CAKUT had renal syndromic HL.
GP group
Among the 105 patients of the GP group, 30 had HL (SNHL:CHL, 24:6). Nineteen of the 24 SNHL patients had genetic or syndromic diseases known to cause SNHL; defective coenzyme Q10 biosynthesis due to COQ6 mutations (n = 9), Alport syndrome (n = 7), Usher syndrome type 1B (n = 1), Fechtner syndrome (n = 1), and mitochondrial myopathy, encephalopathy, lactic acidosis, and stroke-like episodes (n = 1). In the remaining 5 patients, causes of SNHL were multiple congenital anomalies (n = 1), ototoxic chemotherapeutic drug (n = 1), and unknown (n = 3). Among 6 patients with CHL, 5 patients had OME, and 1 patient had cholesteatoma. Two of the 5 patients with OME were diagnosed as having Schimke Immuno-Osseous Dysplasia (SIOD) and 1 patient had Pierson syndrome. Overall, 23 of 30 (76.7%; 20 SNHL and 3 CHL) patients with HL in GP had renal syndromic HL.
CYST group
Among the 39 patients of the CYST group, 5 patients had HL (SNHL:CHL, 2:3). One patient with SNHL had Joubert syndrome and the other had SNHL of unknown cause. All 3 patients with CHL, including 1 patient with Potter syndrome and 1 with NPHP from NPHP1 mutation, had OME. In this group, 3 of 5 (60%; 1 SNHL and 2 CHL) patients with HL had renal syndromic HL.
PP group
Among the 29 patients, 7 patients had HL (SNHL:CHL, 6:1). Six patients with SNHL included 3 very preterm babies, 1 late preterm baby, and 2 term babies. They had various combinations of multiple PP, such as very preterm with bronchopulmonary dysplasia and retinopathy of prematurity (n = 3), culture-proven sepsis (n = 3), perinatal asphyxia (n = 2), along with cardiac arrest, or cardiorespiratory instability during patent ductus arteriosus ligation operation, small for GA or maternal syphilis with fetal alcohol syndrome. CHL with OME was found in a full-term baby with birth asphyxia and multiorgan failure. Overall, renal syndromic HL comprised 1/7 (14.3%, SNHL) cases of HL in PP.
Others group
Among the 64 patients of the others group, 9 patients had HL (SNHL:CHL, 5:4). The causes of SNHL were Kearns-Sayre syndrome (n = 1), mitochondrial cytopathy (n = 1), ototoxic chemotherapeutic drug (n = 2), and unknown (n = 1). All 4 patients with CHL had OME. In this group, renal syndromic HL accounted for 2/9 (22.2%, SNHL) cases of HL.
HL risk factors
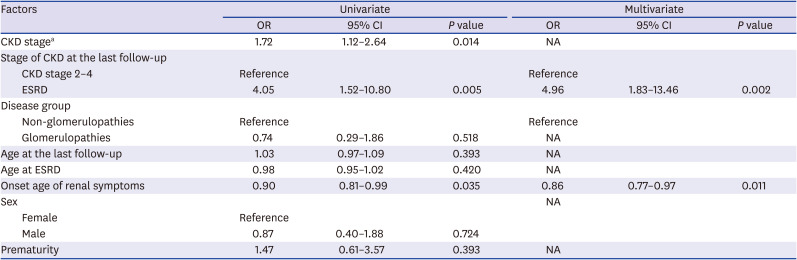
HL increased significantly as the CKD stage increased (OR = 1.43). The risk of HL was significantly higher in ESRD than in CKD stage 2–4 (OR = 2.38) and it remained valid in multivariate analysis. The GP group had a higher risk of HL than the other groups (OR = 2.03). Age, sex, prematurity, age at ESRD, and onset age of renal symptoms did not increase the risk of HL (Table 3). We also analyzed risk factors of SNHL and CHL, respectively. The GP group was the only independent risk factor of SNHL (OR = 3.17) (Table 4). For CHL, the risk increased with the higher CKD stage, ESRD than CKD stage 2–4, and with onset of renal symptoms at a younger age (Table 5).
Table 3
Risk factors of hearing loss
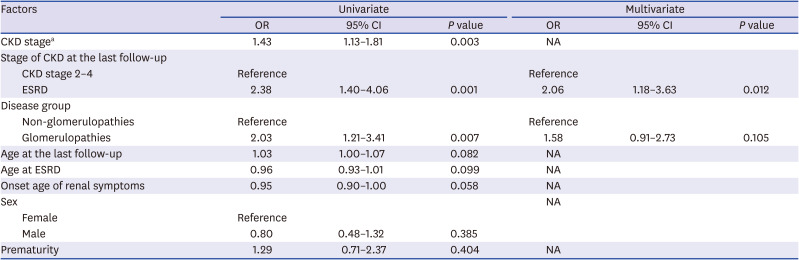
Table 4
Risk factors of sensorineural hearing loss
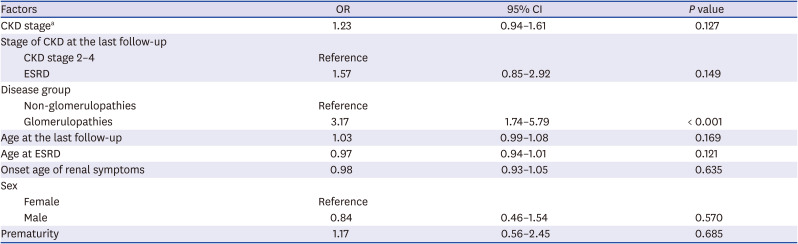
Table 5
Risk factors of conductive hearing loss
DISCUSSION
The overall prevalence of HL in our childhood-onset CKD patients was 19.5%. This prevalence is somewhat lower than in previous reports, 29%–40%,1012 but much higher than that in the general population of over 5%.5 In our study, the overall prevalence of SNHL was 12.1%, which was higher than the 1.2% reported in the Korean adolescents27 (no data available for CHL). Therefore, we suggest that the possibility of HL should be considered in pediatric CKD patients, requiring surveillance and early intervention, if necessary.
Then, which subpopulation of pediatric CKD is at a higher risk of HL? HL was more common in advanced CKD patients in this study, especially in ESRD patients, unlike adult studies that showed no correlation between HL and CKD stage.28 One-fourth of our ESRD patients had HL; CHL was particularly prevalent in ESRD than in non-ESRD patients, probably because of the problem of volume control and the higher susceptibility to infection. On the other hand, sex was not significantly correlated with HL, similar to the findings of previous reports on CKD.1028 Age and follow-up duration were not correlated to HL either. These findings are different from those of an earlier study in the general population, in which male sex and aging were positively related to HL.29 Such differences might reflect the variations in the study population, emphasizing the differences between CKD patients and the general population, or the characteristics of this study of young subjects. In addition to advanced CKD stage, an underlying disease of glomerulopathy was a risk factor of HL, especially SNHL, in this study. This could be attributed to the fact that genetic and syndromic diseases accompanying SNHL were more common in this group, with more than 3/4th of the cases of HL in GP being renal syndromic HL. Meanwhile, prematurity, a significant risk factor for HL in children, did not show significance in this study, in contrast to previous findings showing that the incidences of hearing impairments increased with decreasing GA at birth.19 Nonetheless, the prevalence of HL in our patients with prematurity was 23.0% (17 out of 74), higher than that previously reported in the general preterm population, 0.3%–17.5%.303132 Since congenital or perinatal HL should be detected within three months of birth so that early intervention can begin prior to 6 months of age,3334 their high risk of HL should be recognized for CKD with PP, especially for patients with known risk factors such as small for GA, prenatal asphyxia, or sepsis.1920
Renal syndromic HL was common in this study; half of the HL patients had renal syndromic HL with underlying genetic diseases or syndromic diseases. Approximately half of the patients with SNHL (27/51, 52.9%) had underlying genetic diseases or syndromic diseases well known to cause SNHL, including Alport syndrome, BOR syndrome, and COQ6 mutation. Additionally, four more cases with SNHL classified as renal syndromic HL; Joubert syndrome, where SNHL is not well recognized but was reported previously,35 one case of ACE mutation and two patients with multiple anomalies without a specific diagnosis. In cases of renal tubular dysgenesis, HL is not a typical manifestation; we speculate that SNHL might have developed due to persistent severe hypotension during the neonatal period.36 Among CHL cases, renal syndromic HL was observed in 1/3rd, including Potter syndrome, Pierson syndrome, and SIOD. However, HL has not been reported in association with these syndromes yet, as well as mixed HL in Baller-Gerold syndrome.37 Thus, the causality is uncertain. Nonetheless, these syndromes might make the patients more vulnerable to OME or deter its resolution. Therefore, possibility of HL should be considered in childhood-onset CKD patients, especially those with underlying genetic diseases or syndromic diseases.
As in the general population, OME was present in almost all (27/30, 90.0%) patients with CHL. It is known that children with comorbidities (e.g., Down syndrome or cleft palate) are more commonly affected by OME, and usually more persistently.38 Should CKD be considered such a comorbidity? There was no study of OME comparing the pediatric CKD population with the general population. However, as OME is the one of the most common cause of CHL in infants and young children of the general population7 and we found that younger onset age of renal symptoms and the higher CKD stage are significant risk factors in CHL, we assume that the prevalence of OME might be high in CKD children, especially in those with ESRD, because of the problems with volume control and higher susceptibility to infection, as previously mentioned. Moreover, the spontaneous resolution rate of OME could be lower than that in the general population. Notably, only 7 of 27 cases (25.9%) with repeated audiometry available showed resolution of CHL on follow-up.
This study has several limitations. First, the data for this study were collected in a retrospective manner at a single center. Second, the severity and frequency (high/low) of HL were not addressed. Moreover, the presence of speech impairment and whether it led to speech rehabilitation were not reviewed. Lastly, acquired brain damage or seizure that may affect HL and the use of furosemide, well known as ototoxic drug, were not investigated. Nonetheless, to our knowledge, this is the first paper addressing the prevalence of HL, especially CHL, according to the underlying disease in childhood-onset CKD in a relatively large population of 421 cases. Hearing impairment was found in quite number of pediatric CKD patients, approximately 20%, therefore, we propose that it is important suspect of hearing impairment in this population, thereby detect hearing and speech deterioration in advance so that could lead to early rehabilitation.
In conclusion, approximately 1/5th of our pediatric CKD patients had HL. HL was more common as CKD stage progressed, especially CHL in ESRD, and SNHL was more common in the GP groups than in other groups. Collectively, renal syndromic HL comprised half of the HL cases. SNHL was a manifestation of underlying genetic disease or syndromic disease in more than half of the patients, while CHL was associated with OME in most cases. To improve the quality of life in childhood-onset CKD, we suggest that HL should be considered in pediatric CKD patients, especially those with underlying genetic diseases or syndromic diseases.
 XML Download
XML Download