

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Peroxisomal D-bifunctional protein (DBP), encoded by the HSD17B4 gene, is a peroxisomal enzyme consisting of a hydratase and hydroxyacyl-CoA dehydrogenase (HAD), and catalyzes β-oxidation of very long chain fatty acids (VLCFAs).1 DBP deficiency (MIM #261515) is a rare autosomal recessive Zellweger spectrum disorder (ZSD) caused by biallelic mutations in HSD17B4 located on chromosome 5q23.1, and leads to the accumulation of VLCFAs and deficiency of plasminogen.123 DBP deficiency shares resemblance with clinical features of Zellweger syndrome namely growth failure, neonatal onset encephalopathy, polyneuropathy, hearing and visual impairment, hepatomegaly, and early death in infancy.12 Because peroxisomes are present in all cells and play an important role in cellular functions, including fatty acid oxidation, and phospholipid and bile acid synthesis, patients exhibit clinical and genetic heterogeneity.3 An estimated prevalence of ZSDs and DBP deficiency is 1 in 30,000 and 1 in 100,000, respectively.12 Although patients can be categorized into the neonatal, juvenile, and adult onset DBP deficiency, the onset of symptoms is often in the neonatal period, and present hypotonia (98%) and seizures (93%) in the first month of life.1
Targeted exome sequencing has been an effective diagnostic tool owing to genetic and clinical heterogeneity of ZSDs.4 To date, 83 mutations in HSD17B4 have been reported (April 2019, http://www.hgmd.cf.ac.uk), including 51 missense/nonsense, 10 small indels, 1 gross deletions, 1 gross insertions/duplications, and one rearrangement. DBP deficiency can be classified into 3 types based on the relative impairment of the enzymes activities.12 Type I is a deficiency of both hydratase and HAD units, type II is an isolated hydratase deficiency, and type III is an isolated HAD deficiency. Type I, II, and III represent 27%, 28%, and 45% of the cases, respectively, and exhibited poor prognosis with a mean age at death of 6.9, 10.7, and 17.6 months, respectively.1 Type I patients had deletion, insertion, and nonsense mutations, most type II patients had missense or in-frame mutations in the hydratase domain, and type III patients had mutations in the dehydrogenase domain.12 Recently, Type IV patients presented a missense mutation in each enzyme domain resulting in markedly reduced but detectable hydratase and HAD activities.4
Herein, we report the first Korean neonatal onset DBP deficiency patient with compound heterozygous novel missense mutations in HSD17B4 detected by targeted exome sequencing, and patient's multidisciplinary management for over 2 years.
Go to : 
CASE DESCRIPTION
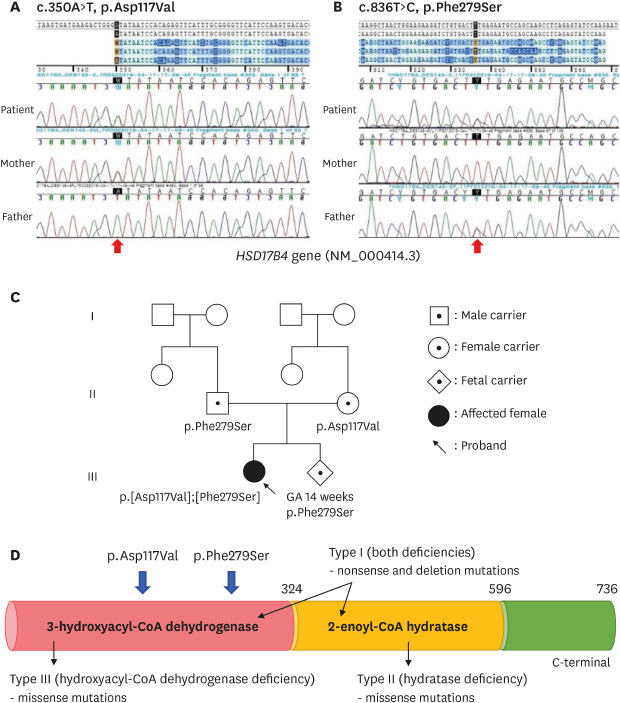
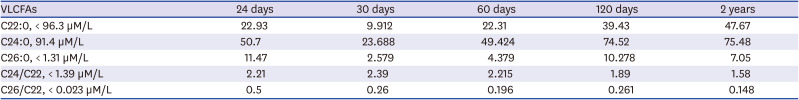
A 28-day-old girl, the first child from non-consanguineous parents, was referred to our hospital owing to hypotonia, neonatal seizure, and elevated serum VLCFAs levels in March 19, 2018. She was born through vaginal delivery at 38+2 weeks, weighed 2.21 kg, and exhibited generalized hypotonia and feeding difficulty at birth and seizure with desaturated oxygen level one day after birth. Although her newborn screening test including tandem MS/MS was normal, metabolic evaluation for neonatal seizure revealed elevated VLCFA levels: C26:0 was 11.47 μmol/L (normal range < 1.31 μmol/L); C24:0/C22:0 was 2.21 (normal range < 1.39), and C26:0/C22:0 was 0.5 (normal range < 0.023) (Table 1). The phytanic and pristanic acid levels at 15 days of age was not detectable. On physical examination, this subject had a high forehead, large anterior fontanel, midfacial hypoplasia, high-arched palate, low-set ear, and clubfoot (Fig. 1). Thus, ZSD was suspected and targeted exome sequencing was performed for several candidate genes. The compound heterozygous mutations c.350A>T (p.Asp117Val) and c.836T>C (p.Phe279Ser), located in representative of 3-hydroxyacyl-CoA dehydrogenase deficiency in HSD17B4 (NM_000414.3) (Fig. 2A and B), were identified and inherited from the mother and father, respectively (Fig. 2C). The allele frequency of c.350A>T was 0.000109 in East Asian and c.836T>C have not reported yet (https://gnomad.broadinstitute.org/). These variants were predicted pathogenic variants by in-silico analysis using PolyPhen-2, Sorting Intolerant From Tolerant, and MutationTaster and classified as likely pathogenic. Although we could not perform the enzyme analysis for 3-HAD and 2-enoyl-CoA hydratase, we assessed the patient as a type III DBP deficiency through the genotype of HSD17B4 (Fig. 2D).
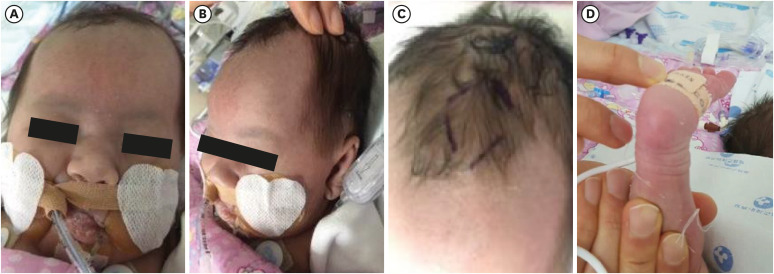 | Fig. 1Clinical photographs of patient. Patient showed (A) typical box-shaped face, (B) large fontanelle, (C) midfacial hypoplasia, and (D) club foot. The figures are published with agreement of parents.
|
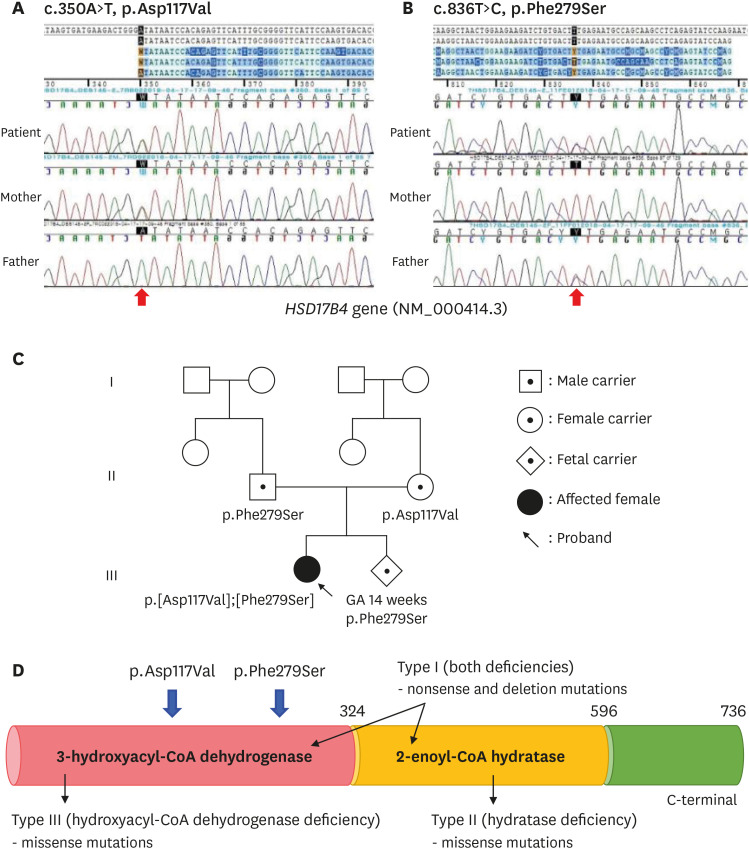 | Fig. 2Partial genomic DNA sequences of HSD17B4 (NM_000414.3) of patients and parents. The result showed compound heterozygote mutation of (A) c.350A>T (p.Asp117Val) and (B) c.836T>C (p.Phe279Ser), and each mutation was inherited from subject parents. (C) Pedigree of patients with D-bifunctional protein deficiency and (D) the locus of two variants representative of 3-hydroxyacyl-CoA dehydrogenase deficiency.
|
Table 1
Chronogical analysis of VLCFAs in the patient with D-bifunctional protein deficiency
![]()
She received a tracheostomy for mechanical ventilation at 35 days of age. Docosahexaenoic acid (DHA) therapy was initiated (100 mg/kg/day), and non-soluble vitamins (A, E, and D) were supplemented. Her adrenal function was normal. The clubfoot was corrected by cast followed by splint application for three months. At 1 month of age, the auditory brainstem response audiometry was performed, and the threshold was 40 dB and 60 dB in the right and left ear, respectively; however, follow-up at 6 months of age showed 90 dB and no response in the right and left ear, respectively. At 2 months of age, the ophthalmologic exam was not specific; however, she showed no eye contact and had paroxysmal asymmetric down beat nystagmus. At 18 months of age, no response to the light reflex in both eyes, optic atrophy, and retinal dystrophy was observed. At 3 days of age, the brain magnetic resonance imaging (MRI) showed the polymicrogyria at bilateral frontal and sylvian area and subependymal cyst at both caudothalamic groove (Fig. 3A and B). The follow-up brain MRI, at 18 months of age, revealed the diffuse atrophy of the cerebral hemispheres, midbrain, and cerebellum and consequent dilatation of third and lateral ventricles (Fig. 3C and D). Moreover, leukodystrophy including diffuse increased T2 signal intensity of bilateral posterior limb of internal capsule, thalami, periventricular white matter, dentate nucleus, corticospinal tract, pons, and relatively sparing subcortical white matter was noted (Fig. 3). She required multiple antiepileptic drugs including clonazepam, zonsisamide, phenobarbital, and valproic acid. At one month of age, EEG revealed slow and disorganized background rhythms and frequent runs of repetitive generalized sharp and slow wave discharges, which were more prominent in the right inferior frontal areas. At 12 months of age, EGG showed slow and disorganized background rhythms and frequent multifocal epileptiform discharges.
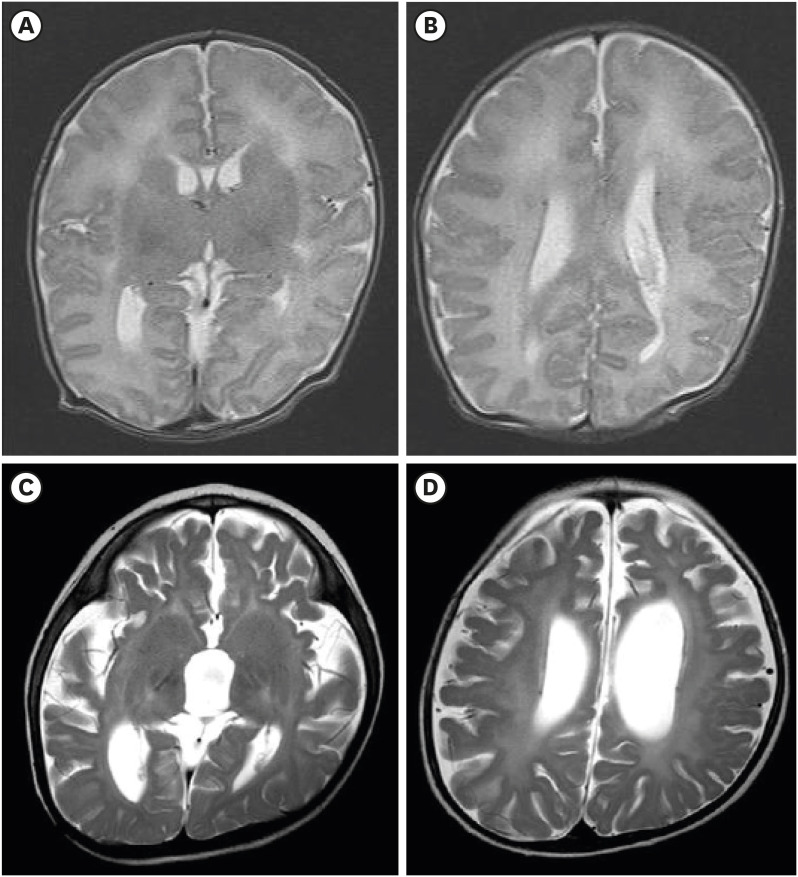 | Fig. 3Brain MRI of the patient. (A, B) Axial T2-image showing bilateral frontal and sylvian polymicrogyria. (C, D) Follow-up images of brain MRI of the patient showing diffuse increased T2 signal intensity and the atrophy of the brain. The patient's parents provided written informed consent for publication of the patient and family information.MRI = magnetic resonance imaging.
|
At 19 months of age, she showed recurrent syndrome of inappropriate antidiuretic hormone secretion (SIADH) which required admission. The serum sodium level decreased to 118 mmEq/L. Urine and serum osmolality was 396 and 247 mOsmol/kg, respectively. The urine sodium, potassium, and chloride concentration was 99.5, 22.4, and 100.1 mEq/L, respectively. The thyroid function test remained normal (thyroid stimulating hormone 1.2 µIU/mL, free T4 0.76 ng/dL). The adrenocorticotropic hormone (ACTH) stimulation test indicated high ACTH (681–760.5 pg/mL) and normal cortisol (19.79–23.29 µg/dL) levels. Thus, hydrocortisone therapy was initiated (8 mg/m2/day). The aldosterone and renin levels were 10.2 ng/dL (normal range, 3.5–24 ng/dL) and 17.3 ng/mL/hr (normal range, 0.3–1.8 ng/mL/hr), respectively.
Our patient's parents had a second baby and were referred to another institution for prenatal genetic testing. The results from chorionic villus sampling revealed the p.Phe279Ser mutation (Fig. 2C). This subject was regularly followed-up in an outpatient clinic although the development did not improve. At 25 months of age, her height, weight, and head circumference was 85.4 cm (75–90 th/p), 11.6 kg (75–90th/p), and 47 cm (50–75th/p), respectively. The serum level of C26:0 seemed to increase with age (Table 1). Home ventilation and gastric tube feeding was continued, and no cholestasis was noted.
Ethics statement
The patient's parents provided written informed consent for publication of the patient and family information.
Go to :
DISCUSSION
Since first described by Watkins et al. in 1989,5 there have been very few reports about DBP deficiency and its management.16789101112 Our patient was brought to the medical attention due to generalized hypotonia, clubfoot, and craniofacial dysmorphism at birth. Although chromosome microarray is considered a first-tier test for such symptoms,13 metabolic work-up including serum VLCFAs must be included for distinguishing the inborn error metabolism which show a facial dysmorphism and neurologic progression.1415 Recently, VLCFAs have been introduced in the newborn screening for detecting X-linked adrenoleukodystrophy (ALD) and other peroxisomal disorders because early-detected X-linked ALD patients can be treated with hematopoietic stem cell transplantation (HSCT), besides cortisol, for progressive brain involvement at early stages.16 For early-detected DBP deficiency patients, the efficacy of HSCT as a treatment option for preventing neurologic deterioration must be assessed, although, to date, there are no reports about HSCT in patients with DBP deficiency.
DBP is also involved in β-oxidation of long-chain polyunsaturated fatty acids, important substrates for DHA biosynthesis, therefore, its deficiency leads to DHA deficiency which affects the brain and retina.3 Although DHA supplementation can increase DHA levels, it cannot improve clinical outcome in patients with DHA deficiency.17 Our patient also showed progressive visual impairment and deteriorating brain despite of early DHA supplementation (within one month).
Recent reports showed early diagnosis of children with DBP deficiency using clinical exome sequencing despite their ambiguous manifestations.9101112 Genotypes including missense, nonsense, or deletion mutations and locations of exon could help predict the prognosis of the patient.2 Our patient had missense mutations located in exons 6 and 12 and the DBP type III phenotype. Since exons 1 to 12 in the DBP gene encode 3-HAD and exons 13 to 19 2-enoyl-CoA hydratase, the deficient enzyme could be predicted using genetic analysis (Fig. 2D).2 All patients presented similar clinical manifestations regardless of their genotype, however, their life span differed. Almost all infants with DBP type I could not survive for more than one year, whereas patients with DBP types II and III often survived for 10 years.1 Our patients also survived for two years and required the multidisciplinary support.
We reported the first Korean neonatal onset DBP deficiency with novel mutations in HSD17B4 and clinical course over 2 years. Our case provided not only the molecular and clinical characteristics of the rare DPB deficiency but also its clinical courses supported by multidisciplinary management, providing evidence that international consensus of ultimate and preventive management for patients with DBP deficiency is warranted.
Go to :
 XML Download
XML Download