

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Although the underlying pathophysiology responsible for diabetic kidney disease (DKD) remains to be fully elucidated, oxidative stress induced by chronic hyperglycemia is increasingly recognized as a significant mechanistic contributor to the development of diabetic complications, including chronic kidney disease and ultimately end-stage renal disease.123 Preclinical and clinical studies have demonstrated that increased oxidative stress enhances the inflammatory response, endoplasmic reticulum (ER) stress, and cell apoptosis in DKD.1456
Some of the oxidative pathways are retained in the mitochondria.7 In particular, nonphagocytic nicotinamide adenine dinucleotide phosphate (NADPH) oxidase 4 (Nox4) is a major source of reactive oxygen species (ROS) in many cell types and the kidney tissue of diabetic animals.8 Recent evidence suggests that Nox4 is located in the intracellular membranes of cardiac myocytes, the nucleus in vascular endothelial cells, the ER in human endothelial cells, and mitochondria in renal cells.7910111213 Our previous experiments showed low mitochondrial content and increased mitochondrial fragmentation in renal tubular cells under high-glucose conditions and the kidneys of diabetic mice.1415 Additionally, we reported that chloroquine (CQ), which has been used as an anti-malarial or anti-inflammatory drug, and amodiaquine (AQ), a derivative of CQ, affects DKD by improving mitochondrial abnormalities.14
We wanted to demonstrate that CQ and AQ reduced renal cell injury under high-glucose conditions by inhibiting mitochondrial Nox4 and reducing ER stress in human renal proximal tubular cells (hRPTCs) and the kidneys of streptozotocin (STZ)-induced diabetic mice.
METHODS
Cell culture
The human kidney cell line (HKC-8, hRPTCs) was provided Dr. L. Rausen (Johns Hopkins University, Baltimore, MD, USA) and was maintained in DMEM/F12 (Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum and 1% penicillin/streptomycin (WelGENE, Daegu, Korea). For the cell studies, HKC-8 cells were cultured with 5 or 30 mM D-glucose for 24 hours. To investigate the effects of the drugs on high glucose-induced HKC-8 cells, CQ (200 μM; Sigma-Aldrich, St. Louis, MO, USA) or AQ (10 μM; Sigma-Aldrich) was added for one hour before changing the culture medium to 30 mM D-glucose, and the cells were collected for analysis. Each cell experiment was performed in triplicate.
Preparation of mitochondrial and subcellular fractions
hRPTCs (3 × 106) were cultured and fractionated into cytosolic (Cyt), mitochondrial, and nuclear fractions using a cell fractionation kit (Abcam, Inc., Cambridge, UK) according to the manufacturer's instructions. Trypsinized HKC-8 cells and medium were collected and centrifuged for 5 minutes at 300 × g. The collected cells were washed twice with buffer A (wash buffer). Then, the cells were counted and resuspended in buffer A to 7 × 106 cells/mL. An equal volume of buffer B (lysis buffer for Cyt extraction) was added to the cell suspensions, mixed via pipetting, and then incubated for 7 minutes at room temperature (RT). The cell lysates were centrifuged at 5,000 × g for 1 minute at 4°C, and the cytosol fractions (cell lysate supernatants) were collected in new tubes. Next, the pellets were resuspended in buffer A, mixed with an equal volume of buffer C (lysis buffer for mitochondrial extraction) via pipetting, and incubated for 10 minutes at RT. The cell lysates were centrifuged at 5,000 × g for 1 minute at 4°C, and the supernatants and pellets were saved. The supernatants were centrifuged at 10,000 × g for 1 minute at 4°C, and the final supernatants were the mitochondrial fractions. The remaining pellets containing the nuclear protein fraction were resuspended in buffer A.
Animal experiments
Eight-week-old male C57BL/6J mice (Center for Research Animals, Seoul, Korea) were used in this study. Diabetes was induced by STZ (Sigma Chemical Co.) through intraperitoneal injection at a dose of 50 mg/kg for five consecutive days. In the experiment, the following four groups of mice (n = 5 in each) were used for three separate experiments: 1) normal control, 2) diabetic control (saline only), 3) diabetes + CQ (50 mg/kg), and 4) diabetes + AQ (20 mg/kg). CQ and AQ were dissolved in saline and used to treat the mice at the indicated doses via intraperitoneal injection at 48-hour intervals for 14 weeks beginning two weeks after STZ injection. All mice were euthanized 16 weeks after STZ administration, and their kidney tissues were collected for analysis. During the experiments, body weights and serum glucose concentrations were measured weekly.
Western blot analysis
hRPTCs and renal tissues were washed with phosphate-buffered saline (PBS) and lysed in ice-cold lysis buffer containing a protease inhibitor (Roche Diagnostics, Mannheim, Germany). The proteins were separated on a 10% Tris-glycine sodium dodecyl sulfate-polyacrylamide gel and then transferred onto a polyvinylidene difluoride (Millipore, Madrid, Spain) membrane using a trans-blot system (Bio-Rad, Hercules, CA, USA). The experiments were performed according to the manufacturer's protocols. Briefly, to prevent non-specific protein binding, the membrane was incubated in 5% bovine serum albumin (BSA) at RT for 1 hour. The membrane was then incubated with primary anti-nitric oxide synthase 2, anti-Bax, anti-cytochrome C (1:1,000; Santa Cruz Biotechnology, Dallas, TX, USA), anti-interleukin (IL)-6 (R&D Systems Inc., Minneapolis, MN, USA), anti-phospho-phosphorylated eukaryotic translation initiation factor 2α (eIF2α), anti-IκB-α, anti-phospho-nuclear factor (NF)-κB, anti-Bcl2 (1:1,000; Cell Signaling Technology, Danvers, MA, USA), anti-Nox4, anti-glucose-regulated protein 78, anti-tumor necrosis factor (TNF)-α, anti-heat shock protein 90, and anti-ptohibitin (1:1,000; Abcam, Inc.) antibodies at 4°C overnight. After washing, the membrane was incubated with horseradish peroxidase (HRP)-conjugated secondary antibody (goat anti-mouse or rabbit-immunoglobulin G-HRP, Santa Cruz Biotechnology) at a ratio of 1:2,000. The protein bands were visualized with enhanced chemiluminescence reagent (BioFX Laboratories, Inc., Owings Mills, MD, USA). Glyceraldehyde 3-phosphate dehydrogenase or β-actin (1:2,000; Santa Cruz Biotechnology) was used as an internal control.
Immunofluorescence
hRPTCs were fixed with 4% paraformaldehyde (PFA), permeabilized, and blocked with BSA. Renal tissue samples were fixed with 4% PFA, dehydrated in ethanol, and embedded in paraffin blocks. Paraffin blocks were cut into 4-µm-thick sections for histological analysis. The samples were incubated with the appropriate primary antibodies. After washing and blocking with PBS, the cells and tissues were re-incubated with secondary antibodies conjugated with Alexa Fluor 488 or 594 (Life Technologies, Seoul, Korea). The samples were counterstained with 4′,6-diamidino-2-phenylindole to delineate the nuclei and examined by confocal microscopy (LSM-700; Carl Zeiss, Jena, Germany).
Measurement of ROS
To determine intracellular and mitochondrial ROS production, the cells were incubated with H2-DCFDA and MitoSOX (Life Technologies) according to the manufacturer's instructions and then examined by confocal microscopy (LSM-700; Carl Zeiss).
Immunohistochemistry
Four-micron-thick kidney sections were rehydrated, and an antigen retrieval procedure was then performed for 30 minutes. Endogenous peroxidase activity was terminated by incubating the tissues with hydrogen peroxide for 5 minutes. The sections were incubated with 8-hydroxy-2′-deoxyguanosine (8-OHdG, 1:100; JaICA, Nikken SEIL Co., Ltd., Shizuoka, Japan) and anti-Nox4 (1:1,000; Abcam, Inc.) antibodies using a biotin-free polymeric HRP-linked antibody conjugate system (Vision BioSystems, Hingham, MA, USA).
Statistics
The data are displayed as the mean ± standard error of the mean. Comparisons between the groups were made with one-way analysis of variance, followed by the Student–Newman–Keuls test. A P value of < 0.05 was considered statistically significant. All of the analyses were completed using GraphPad Prism software (version 5; GraphPad Software, San Diego, CA, USA) for Windows.
Ethics statement
All procedures for the care and use of laboratory animals were approved by the Korea National Institutes of Health. All the methods were performed in accordance with the guidelines of the Animal Research Ethics Committee of Kyung Hee University and the Institutional Animal Care and Use Committee of the Hospital of Kyung Hee University at Gangdong, Seoul, Korea (approval No. KHNMC AP 2017-010).
RESULTS
CQ and AQ inhibited mitochondrial Nox4 and increased mitochondrial mass in hRPTCs cultured in high-glucose concentrations
First, we examined the effects of CQ or AQ on Nox4 activity in renal proximal tubular cells cultured in high-glucose concentrations. Nox4 expression increased in hRPTCs under high-glucose conditions. However, treatment with CQ or AQ abolished high-glucose-induced Nox4 activation in the hRPTCs (Fig. 1A and B, Supplementary Fig. 1). The activity of Nox4 was suppressed after treatment with 200 μM of CQ and 10 μM of AQ (Supplementary Fig. 1). CQ and AQ did not effect cell viability at concentrations from 1 to 400 μM (Supplementary Fig. 2). Functional mitochondrial mass with intact membrane potential, which was indicated by MitoTracker Red staining, decreased under high-glucose concentrations, while treatment with CQ or AQ reversed this effect in the hRPTCs (Fig. 1A). Subcellular fractionation analysis showed that mitochondrial Nox4 was activated by high-glucose concentrations, but CQ and AQ suppressed this activation in hRPTCs, even under high-glucose conditions (Fig. 1C).
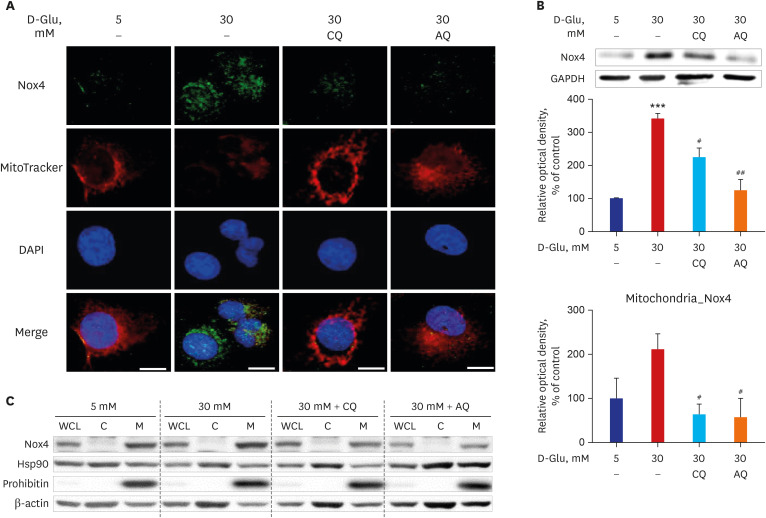
Fig. 1
CQ and AQ inhibited mitochondrial Nox4 and increased mitochondrial mass in hRPTCs under HG conditions. (A) Representative confocal fluorescence images of MitoTracker and Nox4 showing that increased numbers of functioning mitochondria and decreased Nox4 activity were present in hRPTCs after treatment with CQ or AQ under HG conditions. (B) Western blot analysis showed that Nox4 expression was reduced in hRPTCs treated with CQ or AQ under HG conditions. (C) Subcellular fractionation of hRPTCs demonstrated mitochondrial Nox4 activation under HG conditions, but CQ and AQ decreased this activation.
CQ = chloroquine, AQ = amodiaquine, Nox4 = nicotinamide adenine dinucleotide phosphate oxidase 4, DAPI = 4′,6-diamidino-2-phenylindole, GAPDH = glyceraldehyde 3-phosphate dehydrogenase, hRPTC = human renal proximal tubular cell, HG = high-glucose, WCL = whole-cell lysate, C = cytosol, M = mitochondria.
***P < 0.01 vs. 5 mM, #P < 0.05, ##P < 0.01 vs. 30 mM.
![]()
Previously, we reported that high-glucose conditions induced mitochondrial fragmentation and reduced mitochondrial biogenesis in renal proximal tubular cells.1416 Our new findings suggest that a low functional mitochondrial mass caused by high-glucose concentrations was associated with high mitochondrial Nox4 activity, but CQ and AQ successfully ameliorated these mitochondrial changes in hRPTCs under high-glucose conditions.
CQ and AQ reduced mitochondrial ROS generation and ER stress in renal proximal tubular epithelial cells (RPTCs) under high-glucose conditions
The ER forms a membranous network throughout the cytosol and plays roles in protein synthesis, folding, and maturation; Ca2+ storage; and lipid biosynthesis.17 When the ER function is stressed, the unfolded protein response (UPR) is initiated.1718 ER stress has been linked to diabetic complications, including nephropathy.18
We investigated mitochondrial ROS production to identify the effect of healthy mitochondrial mass restoration and Nox4 activity suppression by CQ or AQ in HKC-8 cells under high-glucose conditions. MitoSOX™ Red is a fluorogenic dye for the highly selective detection of superoxide, which is generated in the mitochondria of live cells. Deacetylated H2-DCFDA becomes a fluorescent compound after oxidation by ROS in cells. Fig. 2A shows that CQ and AQ substantially decreased mitochondrial and Cyt ROS generation, as assessed by MitoSOX™ Red and H2-DCFDA staining in HKC-8 cells under high-glucose conditions.
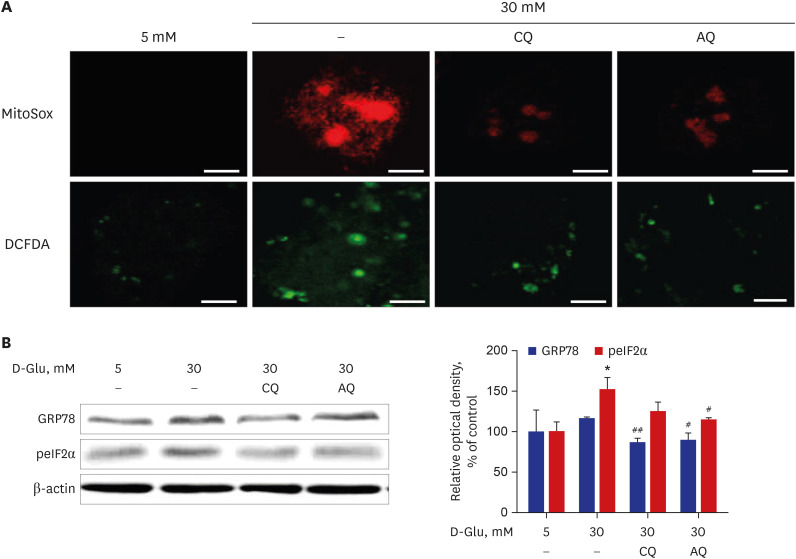
Fig. 2
Effect of CQ and AQ on ROS production and ER stress in hRPTCs under HG conditions. (A) Representative confocal images of MitoSox (red) and H2-DCFDA (green) showing that mitochondrial and intracellular ROS production was reduced in CQ- and AQ-treated hRPTCs under HG conditions. (B) Western blot analyses revealed that the expression of the ER chaperone protein GRP78 was decreased, along with reduced peIF2α after CQ or AQ treatment, even under HG conditions (scale bar: 10 µm).
CQ = chloroquine, AQ = amodiaquine, GRP78 = glucose-regulated protein 78, peIF2α = phosphorylated eukaryotic translation initiation factor 2α, ROS = reactive oxygen species, ER = endoplasmic reticulum, hRPTCs = human renal proximal tubular cells, HG = high-glucose.
*P < 0.05 vs. 5 mM, #P < 0.05, ##P < 0.01 vs. 30 mM.
![]()
Immunoblot analysis suggested that the above reductions in mitochondrial ROS generation were concomitant with lower expressions of G-protein coupled receptor 78 (GPR78) and phospho-eIF2α, which are markers of UPR and ER stress, in hRPTCs incubated with CQ or AQ than in cells treated without CQ or AQ under high-glucose conditions (Fig. 2B).
Taken together, the above results reveal that the repression of Nox4 activity by CQ and AQ treatment contributed to low mitochondrial ROS formation and prevented ER stress in hRPTCs cultured in high-glucose concentrations.
CQ and AQ suppressed inflammatory protein expression and apoptosis in hRPTCs exposed to high-glucose conditions
ER stress causes proinflammatory pathway activation, and if prolonged, this activation can lead to cell damage and apoptotic cell death.19 CQ and AQ downregulated the expression of NF-κB, which is a well-known rapid responder to harmful cellular stimuli, and upregulated the expression of IκB-α in hRPTCs exposed to high-glucose concentrations (Fig. 3A). In addition, treating hRPTCs with CQ and AQ decreased NOS2, IL-6, and TNF-α expression, despite exposure to high-glucose media (Fig. 3B). In cells treated with CQ and AQ, the expression levels of the apoptotic proteins Bax and Cyt C were decreased, whereas the expression level of the anti-apoptotic regulator Bcl2 was restored, even when cultured in high-glucose media (Fig. 3C).
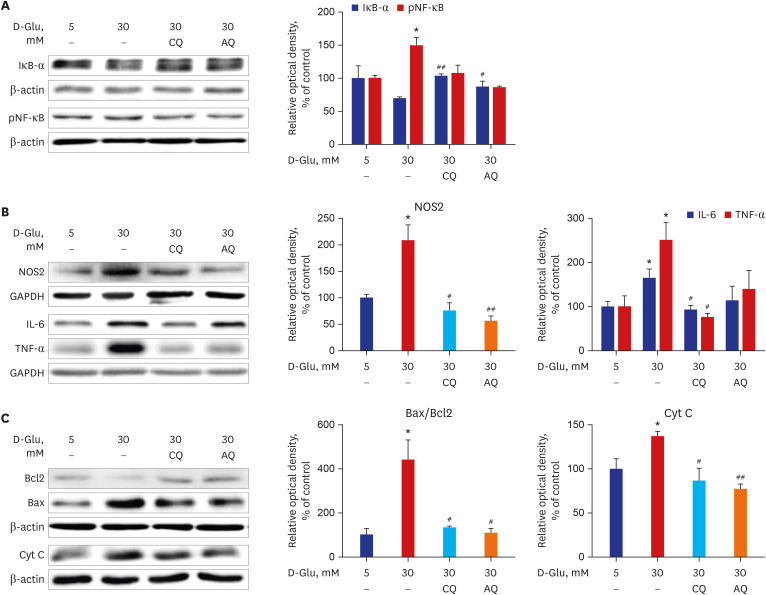
Fig. 3
CQ and AQ inhibited inflammatory protein expression and apoptosis in hRPTCs under HG conditions. (A) Representative immunoblot data showed that IκB-α expression was enhanced but that NF-κB phosphorylation was inhibited in hRPTCs under HG conditions and CQ or AQ treatment. (B) Representative immunoblot data showed decreased protein expression of NOS2, IL-6, and TNF-α after treatment with CQ or AQ under HG conditions. (C) Western blot analyses revealed that the expression levels of the apoptogenic proteins Cyt C and Bax were decreased, while the expression level of the antiapoptotic protein Bcl2 was increased after CQ and AQ treatment under HG conditions. The results are presented as the means ± standard error of the mean for experiments in triplicate.
CQ = chloroquine, AQ = amodiaquine, hRPTCs = human renal proximal tubular cells, HG = high-glucose, pNF-κB = phosphorylated nuclear factor-κB, NOS2 = nitric oxide synthase 2, GAPDH = glyceraldehyde 3-phosphate dehydrogenase, IL = interleukin, TNF = tumor necrosis factor, Cyt = cytosolic.
*P < 0.05 vs. 5 mM, #P < 0.05, ##P < 0.01 vs. 30 mM.
![]()
As illustrated by our data, CQ and AQ attenuated the high-glucose-induced activation of the proinflammatory pathway and the expression of cell apoptosis-associated proteins in RPTCs exposed to high-glucose media.
CQ and AQ treatment attenuated oxidative stress and ER stress in diabetic kidneys
To determine the beneficial effects of CQ and AQ on DKD in vivo, diabetes was induced in mice by STZ injection, and CQ or AQ was administered to the diabetic mice for 14 weeks.
The concentration of 8-OHdG is an indicator of oxidative stress within cells. The immunohistochemical detection of oxidative DNA damage by 8-OHdG indicated that the tubular 8-OHdG content was lower in the kidneys of the drug-treated groups than in the renal tubules of the control group but higher in those of the diabetic control group (Fig. 4A). Western blot analysis and immunohistochemistry using an anti-Nox4 antibody revealed that Nox4 expression was reduced in the kidneys of the diabetic mice in the CQ and AQ-treated groups compared to the kidneys of the diabetic control group (Fig. 4A and B). Fig. 4B shows that CQ and AQ administration ameliorated ER stress in diabetic kidneys and decreased the expression of GPR78 and phospho-eIF2α, markers of the UPR.
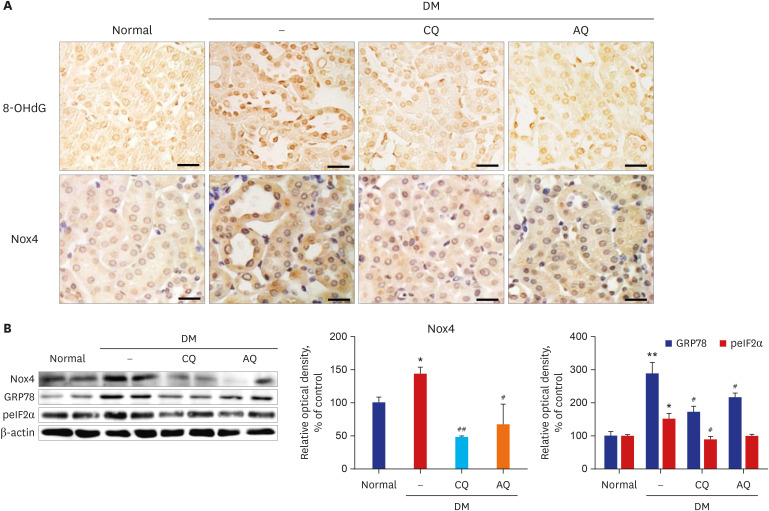
Fig. 4
CQ or AQ treatment attenuated oxidative stress and endoplasmic reticulum stress in STZ-induced diabetic mice. (A) The expression of 8-OHdG and Nox4 was increased in the kidneys of the diabetic group compared to the control group but was reduced by CQ and AQ injection (scale bar: 20 µm). (B) In the diabetic kidneys, Nox4 protein expression was markedly increased. However, CQ or AQ treatment ameliorated the expression of Nox4. (C) CQ or AQ treatment reduced GRP78 and phospho-eIF2α expression in the diabetic kidneys (n = 5 per group).
CQ = chloroquine, AQ = amodiaquine, 8-OHdG =8-hydroxy-2′-deoxyguanosine, Nox4 = nicotinamide adenine dinucleotide phosphate oxidase 4, GRP78 = glucose-regulated protein 78, peIF2α = phosphorylated eukaryotic translation initiation factor 2α, STZ = streptozotocin, NADPH = nicotinamide adenine dinucleotide phosphate, DM = diabetes mellitus.
*P < 0.05, **P < 0.01 vs. normal, #P < 0.05, ##P < 0.01 vs. DM control.
![]()
CQ and AQ treatment diminished inflammatory protein expression and albuminuria in diabetic mice
We hypothesized that these drug-induced reductions in oxidative stress resulted in conditions favorable for inflammation in diabetic kidneys. We examined the expression of inflammatory proteins in the kidneys of normal mice, diabetic mice, and diabetic mice treated with CQ or AQ.
The activation of proinflammatory pathways in diabetic kidneys was accompanied by increased expression of phospho-NF-κB, IL-6, and TNF-α. However, proinflammatory pathway activation was ameliorated in the kidneys of the mice that were administered CQ and AQ (Fig. 5A).
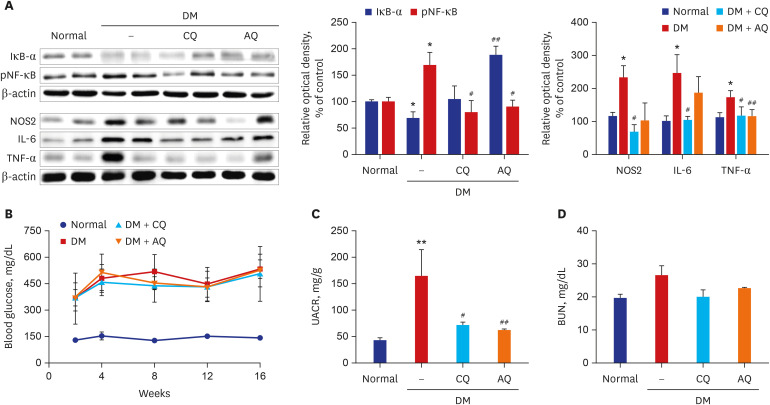
Fig. 5
CQ or AQ treatment attenuate inflammatory protein expression and albuminuria in STZ-induced diabetic mice. (A) In diabetic kidneys, treatment with CQ or AQ increased IκB-α, but reduced other inflammatory proteins including pNF-κB, NOS2, IL-6, and TNF-α. (B) The STZ-induced diabetes group had higher blood glucose levels than the normal control group. (C) However, CQ- and AQ-treated STZ-induced diabetic mice exhibited a significant reduction in the urine albumin excretion rate compared with diabetic control mice. (D) Levels of BUN in four mice groups (n = 5 per group).
DM = diabetes mellitus, CQ = chloroquine, AQ = amodiaquine, pNF-κB = phosphorylated nuclear factor-κB, NOS2 = nitric oxide synthase 2, IL = interleukin, TNF = tumor necrosis factor, STZ = streptozotocin, BUN = blood urea nitrogen.
*P < 0.05, **P < 0.01 vs. normal, #P < 0.05, ##P < 0.01 vs. DM control.
![]()
Notably, CQ and AQ significantly reduced urine albumin amounts in diabetic mice without significant changes in blood sugar levels (Fig. 5B and C). There were no significant differences in the blood urea nitrogen levels between the four groups of mice (Fig. 5D).
DISCUSSION
An increasing body of evidence has shown that renal tubules play roles as significant as renal glomeruli in the pathogenesis of DKD.20 Moreover, recent experimental studies have revealed that the altered processing of filtered albumin by renal tubules is critical for the development of albuminuria in early DKD.2122 Renal tubular cells have ample mitochondria to meet the continuous ATP requirements needed to facilitate their resorptive function.16 Here, we showed that CQ and AQ reduced the activation of mitochondrial Nox4 and suppressed the expression of proinflammatory and UPR-associated proteins in diabetic kidneys and RPTCs exposed to high-glucose media. Both CQ and AQ improved diabetic tubulopathy, which changed along with decreases in albuminuria in STZ-induced diabetic mice.
Mitochondria are small intracellular organelles that produce energy and are formation sites of radicals and other ROS in cells.232425 Recently, abnormal mitochondria or mitochondrial dysfunction has emerged as an important pathogenesis mechanism of DKD.1416262728 Dysmorphic mitochondria have been reported in the proximal renal tubular cells of type 2 diabetic patients with microalbuminuria.29 Frequent mitochondrial DNA deletions and reductions in mitochondrial protein expression have also been reported in the kidneys of diabetic mice.30 In addition, metabolomic analysis of the urine of patients with DKD supports the hypothesis that mitochondrial dysfunction is crucial in this disease.27 We also substantiated that the reversal of abnormal mitochondrial dynamics and quality control in renal proximal tubular cells exposed to high-glucose conditions attenuated diabetic tubulopathy.141631 Notably, drug treatments maintaining mitochondrial homeostasis have been shown to decrease albuminuria and improve tubulointerstitial pathology in STZ-induced diabetic mice.141631
A small quantity of oxidant species is necessary to maintain normal physiology in cells, but there is strong evidence that excessive oxidative stress is implicated in the development of DKD.32 NADPH oxidases are the only known enzymes dedicated solely to ROS generation with their catalytic subunits.33 Human isoforms of the catalytic component of the complex include Nox1–5 and dual oxidase 1–2,34 and the roles of several human isoforms in DKD have already been studied.32 Nagasu et al.35 suggested that increased endothelial Nox2 activity was associated with renal injury in transgenic diabetic Akita mice. However, Nox2-deficient mice did not reveal a role for Nox2 in DKD, despite a reduction in macrophage infiltration.36 Jha et al.1 showed that Nox4 was the main source of renal ROS in a mouse model of diabetic nephropathy. These authors demonstrated that the deletion of Nox4 but not Nox1 reduced glomerular injury accompanied by attenuated albuminuria, preserved structure, and decreased glomerular macrophage infiltration in STZ-induced diabetic ApoE(−/−) mice.1 Kim et al.9 reported that ROS injury mediated by angiotensin II-induced mitochondrial Nox4 played a pivotal role in mitochondrial dysfunction in tubular cells. In our study, CQ and AQ increased the normally functional mitochondria and suppressed the activation of Nox4 located in the mitochondria in renal proximal tubular cells exposed to high-glucose media (Fig. 1). Previously, we reported that these two anti-malarial drugs, CQ and AQ, induced AMP-activated protein kinase (AMPK) α phosphorylation and improved mitochondrial fragmentation.14 Taken together, these observations suggest that increased phospo-AMPKα induced by CQ and AQ ameliorated mitochondrial disorder in renal tubular cells incubated with high-glucose media, resulting in low Nox4 activity in the cells. Consistently, the inhibitory effects of CQ and AQ on Nox4 were observed in diabetic kidneys, in parallel with reductions in albuminuria and oxidative tissue damage (Fig. 4A and B, Fig. 5C).
ER stress occurs in pathological conditions that increase the demand for protein folding or disrupt normal folding processes.37 A large body of evidence has indicated that ER stress is implicated in the development of diabetic nephropathy.38 We also observed increased ER stress in proximal tubular cells incubated with high-glucose media and diabetic kidneys in which GPR78 and phospho-eIF2α expression was increased (Figs. 2C and 4C). GPR78 is an ER chaperone and serves as a master modulator of the UPR signaling network.39 ER stress leads to GPR78 dissociation from protein kinase R-like ER kinase (PERK). Activated PERK by GPR78 dissociation phosphorylates eIF2α, which reduces the protein load on the damaged ER.40 In our study, treatment with CQ and AQ suppressed the expression of Nox4 and ER stress markers (Figs. 2C and 4C). In parallel, CQ and AQ decreased inflammatory markers, such as NF-κB, IL-6, and TNF-α (Fig. 3A and B, Fig. 5B). Persistent ER stress is well known to activate the proinflammatory pathway via NF-κB.19 An interplay between ER and Nox has been proposed, and Nox4 has been reported to play either prosurvival or proapoptotic roles during ER stress.41 In our study, Nox4 suppression by CQ and AQ seemed to contribute to cell survival along with decreased ER stress in diabetic tubulopathy (Fig. 3C).
We induced diabetes in mice by injections of STZ, a chemical toxin for pancreatic β-cells. This model has the advantage of simplicity in producing high serum glucose levels. However, these diabetic mice do not replicate all the features of human diabetic nephropathy as other animal models for DKD.42 In our study, renal function did not decline in the mice, despite persistently high serum glucose levels (Fig. 5B and D). However, it should be cautiously noted that STZ-induced diabetic mice generally represent type 1 diabetes mellitus only due to a lack of insulin resistance.
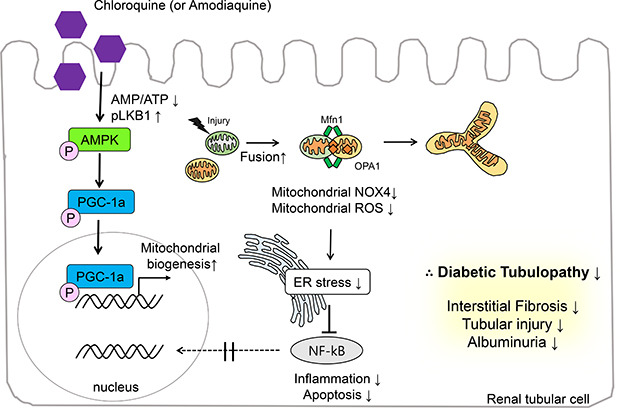
In Fig. 6, we summarized the protective actions of CQ and AQ in renal tubular cells under high-glucose conditions. Our previous report14 showed that both CQ and AQ induced AMPK phosphorylation fueled by a fall in the AMP/ATP ratio and liver kinase B1 phosphorylation. Subsequently, increased phospho-PGC-1α contributes to mitochondrial biogenesis and mitochondrial fusion. In this study, we determined that mitochondrial homeostasis maintained by CQ and AQ was associated with the suppression of mitochondrial Nox4 activity, thereby alleviating ER stress and diabetic tubulopathy. Further studies exploring the roles of mitochondrial Nox4 in the pathogenesis of DKD could suggest new therapeutic targets for patients with DKD.
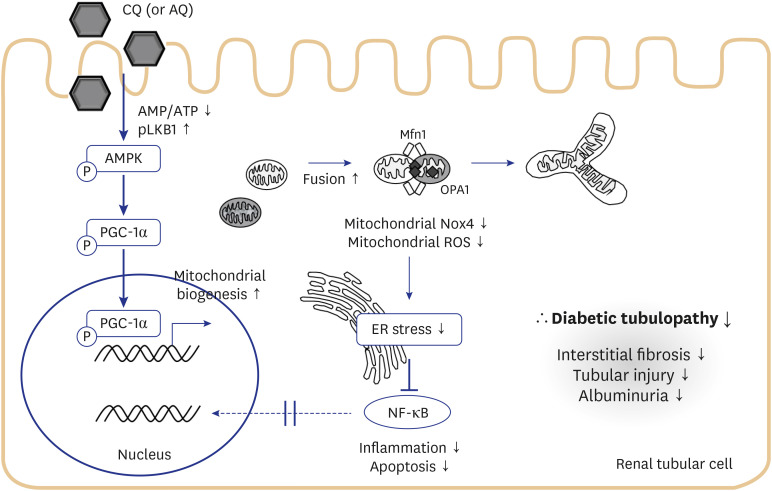
Fig. 6
Schematic representation of the proposed model for the effects of CQ and AQ on diabetic tubulopathy. Our previous report14 suggested that these drugs induce AMPK phosphorylation fueled by a fall in AMP/ATP ratio and liver kinase B1 phosphorylation. Subsequently, increased phospho-PGC-1α contributes to mitochondrial biogenesis and mitochondrial fusion. In this study, we figured out that mitochondrial homeostasis maintained by CQ and AQ is connected with suppression of Nox4 activity, thereby alleviating ER stress, inflammation, and apoptosis. Consequently, we observed decreased tubular injury, interstitial fibrosis in kidneys of diabetic mice after treatment with CQ and AQ concomitant with reduced albuminuria.
AMPK = AMP-activated protein kinase, CQ = chloroquine, AQ = amodiaquine, ER = endoplasmic reticulum, NF-κB = nuclear factor-κB, Nox4 = nonphagocytic nicotinamide adenine dinucleotide phosphate oxidase 4, ROS = reactive oxygen species.
![]()
 XML Download
XML Download