

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Telmisartan is an angiotensin II type 1 receptor blocker (ARB) that inhibits the body's renin-angiotensin-aldosterone system (RAAS) whereby it is widely used to treat hypertensive patients.1 In addition to its inhibitory effects on the RAAS, telmisartan exhibits various ancillary effects including vascular protection effects. In this regard, we reported that telmisartan attenuates vascular inflammation by decreasing tumor necrosis factor α (TNFα) signaling pathway in bovine aortic endothelial cells.2 Most recently, we also reported that telmisartan decreases vascular smooth muscle cell (VSMC) contractility and vessel contraction by activating AMP-activated protein kinase (AMPK)/proteasome/myosin light chain kinase degradation signaling cascade in rat VSMCs and aortas.3
VSMCs comprise a large percentage of the medial layer of the vascular wall, which is continuously exposed to mechanical and/or biochemical stimuli, and they have an important role in the maintenance of vascular homeostasis including vascular tone in response to diverse stressors.4 Therefore, physiological function dysregulation of VSMCs is associated with the development of various vascular diseases, such as hypertension, atherosclerosis, and restenosis.5 In this respect, functional normalization of VSMCs by pharmacological intervention that would inhibit abnormal VSMC proliferation have been considered as a measure for the prevention and treatment of atherosclerosis and post-intervention restenosis.
Although the contribution of abnormal VSMC proliferation to the development of diverse vascular diseases, including atherosclerosis and restenosis, has been identified, the effects of telmisartan on VSMC proliferation and its mechanism of action have not been fully elucidated. In the current study, we investigated the molecular mechanism by which telmisartan inhibits rat VSMC proliferation.
Go to : 
METHODS
Materials
Telmisartan, losartan, and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Cayman Chemicals (Ann Arbor, MI, USA). Fimasartan was a kind gift from Boryung Pharmaceuticals (Seoul, Korea). GW9662 and dimethyl sulfoxide (DMSO) were obtained from Sigma-Aldrich (St. Louis, MO, USA). Compound C was purchased from Calbiochem (Darmstadt, Germany). Recombinant rat platelet-derived growth factor (PDGF)-BB protein was purchased from Novus Biologicals (Centennial, CO, USA). Antibodies against mammalian target of rapamycin (mTOR), p-mTOR-Ser2448, p70 S6 kinase (p70S6K), p-p70S6K-Thr389, AMPK, p-AMPK-Thr172, acetyl-CoA carboxylase (ACC), and p-ACC-Ser79 were purchased from Cell Signaling Technology (Beverly, MA, USA). Antibody against β-actin was purchased from Sigma-Aldrich. Dulbecco's modified Eagle's medium (DMEM) was obtained from Fisher Scientific (Ottawa, Canada). Dulbecco's phosphate-buffered saline (DPBS), fetal bovine serum (FBS), penicillin and streptomycin antibiotics, trypsin–EDTA solution, and plasticware for cell culture were purchased from Gibco-BRL (Gaithersburg, MD, USA). All other chemicals used were of the purest analytical grade available.
Cell culture and drug treatments
Rat aortic VSMCs were isolated and cultured as previously described.3 Cells between passages 3 and 7 were used for all experiments. VSMCs grown to 90% confluence in 60-mm culture dishes were incubated in the absence or presence of various concentrations of telmisartan for 24 hours, or in 40 μM telmisartan for the indicated times, in DMEM supplemented with 2% FBS. In some experiments, cells were co-treated with the indicated drugs or chemicals for the indicated times.
Western blot analyses
After VSMCs were treated with telmisartan in the absence or presence of various chemicals, total proteins were extracted from the cells, and then subjected to western blot analyses, as described previously.3 The primary antibody dilutions used for the western blot analyses were p-mTOR-Ser2448 (1:1,000), mTOR (1:1,000), p-ACC-Ser79 (1:3,000), ACC (1:1,000), p-p70S6K-Thr389 (1:1,000), p70S6K (1:1,000), p-AMPK-Thr172 (1:1,000), AMPK (1:1,000), and β-actin (1:3,000).
Transfection of dominant-negative (dn)-AMPKα1 constructs
The dn-AMPKα1 cDNA construct was a kind gift from Prof. Joohun Ha (Kyung Hee University, Seoul, Korea). Transfection of pcDNA3.1 vector containing rat dn-AMPKα1 cDNA carrying a point mutation of GAC→GCC at 157th codon (D157A mutant) was performed as previously described.3 Briefly, the dn-AMPKα1 cDNA construct was transfected into VSMCs grown to 70% confluence in 60-mm culture dishes, using Lipofectamine 2000 from Invitrogen (Carlsbad, CA, USA), following the manufacturer's instructions. Equal amounts of pcDNA3.1 vector were transfected for control. After incubation for 5 hours at 37°C, the cells were further incubated in DMEM containing 10% FBS for 24 hours before telmisartan treatment.
Measurement of cell proliferation
Cell proliferation was determined by using the MTT assay. VSMCs were seeded into 12-well culture plates in DMEM supplemented with 10% FBS and cultured at 37°C for 24 hours. When the cells reached 50% confluence, the medium was replaced with 2% FBS DMEM containing 40 µM telmisartan. The cells were allowed to proliferate for 24 hours, after which MTT solution (5 mg/mL) was added to each well (1:10), and the cells incubated for a further 2 hours. On completion, the supernatants were aspirated and the wells washed with DPBS once. Formazan was then solubilized with 500 μL DMSO. An aliquot (200 µL) of this solution was transferred to 96-well plates. Cell proliferation was assessed by measuring the absorbance at 570 nm using a microplate reader (Model 680 Microplate Reader, Bio-Rad Laboratory, CA, USA). Each experiment was performed in quadruplicate.
Statistical analyses
All results are expressed as mean ± standard deviation (SD) values, with n indicating the number of experiments. Statistical significance of differences between two mean values was evaluated using Student's t-test. All differences were considered significant at a P value < 0.05.
Go to :
RESULTS
Telmisartan decreases rat VSMC proliferation by inhibiting p-mTOR-Ser2448 and p-p70S6K-Thr389
Although it has been previously reported that prolonged telmisartan treatment (7 days) inhibits proliferation of human aortic and carotid artery smooth muscle cells,6 telmisartan as an antihypertensive agent is treated once daily, and its elimination half-life is approximately 24 hours;7 therefore, telmisartan treatment for 24 hours is compatible with clinical situations. Initially, we investigated whether treatment of rat VSMCs with telmisartan for 24 hours can inhibit cell proliferation. As shown in Fig. 1A, telmisartan inhibited VSMC proliferation in a dose-dependent manner. VSMC proliferation in cells treated with 40 μM telmisartan for 24 hours was approximately 40% lower than that of vehicle control cells (Fig. 1A). We next examined whether telmisartan inhibits the mTOR/p70S6K signaling pathway in rat VSMCs. As expected, telmisartan repressed p-mTOR-Ser2448 and p-p70S6K-Thr389 in dose- and time-dependent manners with no alteration of their total protein expressions (Fig. 1B and C). These results suggested that telmisartan inhibited VSMC proliferation via downregulation of the mTOR/p70S6K signaling pathway.
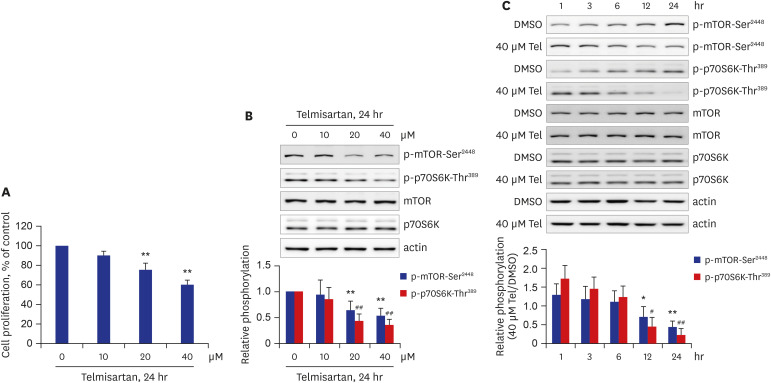 | Fig. 1Telmisartan decreases rat VSMC proliferation by inhibiting p-mTOR-Ser2448 and p-p70S6K-Thr389. (A) VSMC proliferation was measured by MTT assay as described in the METHODS. VSMCs were treated with various doses of telmisartan (0, 10, 20, or 40 μM) for 24 hours. (B) Cells were treated as described above, and then p-mTOR-Ser2448 and p-p70S6K-Thr389 levels were detected using western blot analyses. Nitrocellulose membranes were re-probed with antibodies against mTOR, p70S6K, or β-actin to assess equal sample loading. (C) VSMCs were treated with 40 µM telmisartan or vehicle (DMSO) for the indicated times (0, 1, 3, 6, 12, or 24 hours), and the subsequent p-mTOR-Ser2448 and p-p70S6K-Thr389 levels were detected using western blot analyses as described in Fig. 1B. (B, C) Using western blot results, densitometry was performed to quantitate p-mTOR-Ser2448 and p-p70S6K-Thr389 levels relative to those of total mTOR and p70S6K, respectively. All experiments were performed at least four times independently, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations below the control level (± standard deviation).Differences were considered statistically significant at *P < 0.05, #P < 0.05, **P < 0.01, and ##P < 0.01.
|
Telmisartan-activated AMPK inhibits VSMC proliferation by decreasing mTOR/p70S6K signaling pathway
Next, we investigated whether telmisartan would increase AMPK activity under our experimental conditions. As shown in Fig. 2A and B, AMPK phosphorylation at Thr172 was elevated by telmisartan in dose- and time-dependent manners. The level of p-AMPK-Thr172 was approximately 4-fold higher in VSMCs treated with 40 μM telmisartan for 24 hours, than in vehicle (DMSO) control cells (Fig. 2A). In parallel with the increase in p-AMPK-Thr172, telmisartan also markedly increased p-ACC-Ser79, a well-known downstream effector of AMPK (Fig. 2A and B), further indicating that telmisartan can enhance AMPK activity. To reveal the role of AMPK in telmisartan inhibition of p-mTOR-Ser2448 and p-p70S6K-Thr389, we performed inhibitor-based studies using compound C, a specific AMPK inhibitor. As shown in Fig. 2C, co-treatment with 10 μM compound C completely reversed the telmisartan-induced reductions of p-mTOR-Ser2448 and p-p70S6K-Thr389. As expected, the telmisartan-induced increase in p-ACC-Ser79 was significantly attenuated in compound C-co-treated cells (Fig. 2C), suggesting that compound C sufficiently inhibited AMPK activation. To confirm these results, we introduced dn-AMPKα1 (D157A mutant) constructs into VSMCs. As shown in Fig. 2D, overexpression of the dn-AMPKα1 gene in VSMCs was successful, based on the increase in total AMPK expression and the reductions of p-AMPK-Thr172 and p-ACC-Ser79 in the dn-AMPKα1 gene-transfected cells. Similar to the compound C results, telmisartan-induced p-mTOR-Ser2448 and p-p70S6K-Thr389 inhibitions were significantly restored in dn-AMPKα1-overexpressed VSMCs (Fig. 2E). Furthermore, ectopic expression of the dn-AMPKα1 gene significantly reversed telmisartan-repressed VSMC proliferation (Fig. 2F). These results show that telmisartan-activated AMPK inhibits VSMC proliferation by downregulating the mTOR/p70S6K signaling axis.
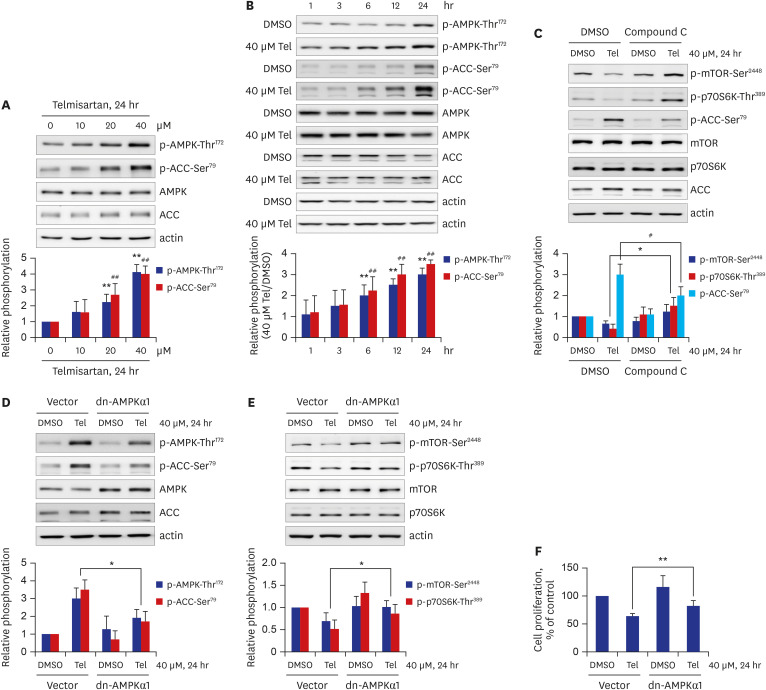 | Fig. 2Telmisartan-activated AMPK inhibits VSMC proliferation by decreasing mTOR/p70S6K signaling pathway. (A) VSMCs were treated with various doses of telmisartan (0, 10, 20, or 40 μM) for 24 hours followed by p-AMPK-Thr172 and p-ACC-Ser79 detection using western blot analyses as described in Fig. 1B. (B) VSMCs were treated with 40 µM telmisartan or vehicle (DMSO) for the indicated times (0, 1, 3, 6, 12, or 24 hours), and the p-AMPK-Thr172 and p-ACC-Ser79 levels were detected using western blot analyses as described in Fig. 1B. (C) VSMCs were treated with 40 µM telmisartan or vehicle (DMSO) in the absence or presence of 10 µM compound C for 24 hours followed by p-mTOR-Ser2448, p-p70S6K-Thr389, and p-ACC-Ser79 detection using western blot analyses as described in Fig. 1B. (D, E) VSMCs were transfected with rat dn-AMPKα1 (D157A) gene or empty vector before treatment with 40 µM telmisartan or vehicle (DMSO) for 24 hours. The resulting p-AMPK-Thr172, p-ACC-Ser79, p-mTOR-Ser2448, and p-p70S6K-Thr389 levels were detected using western blot analyses as described in Fig. 1B. (F) Rat dn-AMPKα1 (D157A) gene- or empty vector-transfected VSMCs were treated with 40 µM telmisartan or vehicle (DMSO) for 24 hours and the subsequent cell proliferation was assessed by MTT assay as described in the METHODS. All experiments were independently performed at least four times, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above/below the control levels (± standard deviation).Differences were considered statistically significant at *P < 0.05, #P < 0.05, **P < 0.01, and ##P < 0.01.
|
Among the tested ARBs, only telmisartan induces p-AMPK-Thr172 and inhibits mTOR/p70S6K signaling pathway and VSMC proliferation
Although it has been well-established that ARBs produce common blood-pressure-lowering effects by blocking the RAAS in the body,8 it has also been recognized that each ARB exerts specific effects on diverse physiological processes, including vascular protection effects.9 This premise led us to examine whether ARBs other than telmisartan could affect AMPK activity. As shown in Fig. 3A, of the ARBs tested (telmisartan, losartan, and fimasartan), only telmisartan treatment produced an increase in p-AMPK-Thr172 and p-ACC-Ser79. Furthermore, p-mTOR-Ser2448 and p-p70S6K-Thr389 levels were only inhibited in telmisartan-treated VSMCs (Fig. 3B). In line with these results, VSMC proliferation was only lowered in telmisartan-treated VSMCs (Fig. 3C). These results show that inhibition of VSMC proliferation by AMPK-mediated downregulation of mTOR/p70S6K signaling pathway is a particular effect of telmisartan, not of losartan or fimasartan.
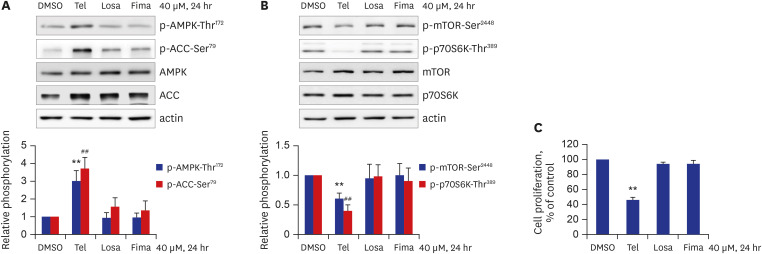 | Fig. 3Among the tested ARBs, only telmisartan induces p-AMPK-Thr172 and inhibits mTOR/p70S6K signaling pathway and VSMC proliferation. (A, B) VSMCs were treated with various ARBs (telmisartan, losartan, or fimasartan; all at a dose of 40 μM) for 24 hours, and then p-AMPK-Thr172, p-ACC-Ser79, p-mTOR-Ser2448, and p-p70S6K-Thr389 levels were assessed using western blot analyses as described in Fig. 1B. (C) VSMCs were treated as described above, and cell proliferation was evaluated by MTT assay as described in the METHODS. All experiments were independently performed at least four times, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above/below the control level (± standard deviation).Differences were considered statistically significant at **P < 0.01 and ##P < 0.01.
|
Telmisartan-inhibited VSMC proliferation is mediated by a PPARγ-independent pathway
In addition to its blockage of angiotensin II type 1 receptor, telmisartan has also been reported to act as a partial peroxisome proliferator-activated receptor γ (PPARγ) agonist.1 Hence, to investigate PPARγ involvement in telmisartan-repressed VSMC proliferation via AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway, we performed inhibitor studies using GW9662, a specific and irreversible PPARγ inhibitor. As shown in Fig. 4A, co-treatment with 5 μM GW9662 did not affect telmisartan-augmented p-AMPK-Thr172 and p-ACC-Ser79. Similarly, p-mTOR-Ser2448 and p-p70S6K-Thr389 levels remained unchanged after GW9662 co-treatment (Fig. 4B). In agreement with these results, telmisartan-inhibited VSMC proliferation was unaffected by co-treatment with 5 μM GW9662 (Fig. 4C). These results suggest that PPARγ is not involved in the telmisartan-induced decrease in VSMC proliferation.
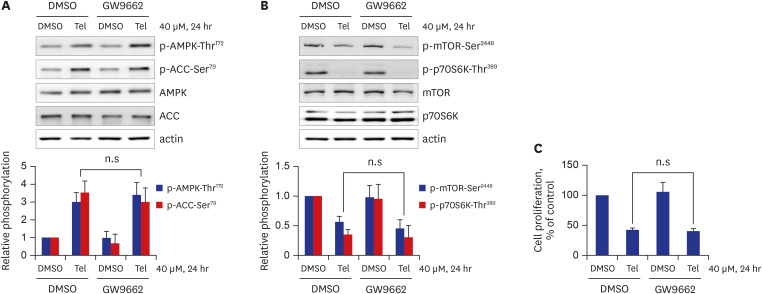 | Fig. 4Telmisartan-inhibited VSMC proliferation is mediated by a PPARγ-independent pathway. (A, B) VSMCs were treated with 40 µM telmisartan or vehicle (DMSO) in the absence or presence of 5 µM GW9662 for 24 hours, and subsequent p-AMPK-Thr172, p-ACC-Ser79, p-mTOR-Ser2448, and p-p70S6K-Thr389 levels were determined using western blot analyses as described in Fig. 1B. (C) VSMCs were treated as described above, and cell proliferation was measured by MTT assay as described in the METHODS. All experiments were independently performed at least four times, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above/below the control level (± standard deviation).n.s = not significant.
|
Telmisartan inhibits PDGF-stimulated VSMC proliferation through AMPK-mediated inhibition of mTOR/p70S6K signaling pathway
The growth factor PDGF has been reported to be responsible for the development of various vascular diseases, including atherosclerosis.10 PDGF can stimulate aberrant VSMC proliferation, ultimately resulting in vascular atherosclerosis.1011 Thus, treatment of cells with PDGF is commonly used to mimic in vitro atherosclerosis.1112 Therefore, we co-treated VSMCs with telmisartan and 10 ng/mL PDGF to investigate whether the anti-proliferative effects of telmisartan occur in a PDGF-stimulated in vitro atherosclerosis model system. As expected, VSMC proliferation was increased by approximately 1.3-fold in PDGF-stimulated cells compared to that in PDGF-untreated VSMCs, and co-treatment with telmisartan inhibited the PDGF-stimulated VSMC proliferation in a dose-dependent manner (Fig. 5A). Furthermore, in parallel with results observed under PDGF-untreated conditions (Fig. 1B), telmisartan dose-dependently decreased p-mTOR-Ser2448 and p-p70S6K-Thr389 levels in PDGF-stimulated VSMCs, but there was no alteration of the total protein expressions (Fig. 5B). Similar to the results obtained in PDGF-unstimulated VSMCs (Fig. 2A), telmisartan dose-dependently increased p-AMPK-Thr172 and p-ACC-Ser79 levels in PDGF-treated VSMCs (Fig. 5C). Next, to confirm the role of AMPK activity in the telmisartan inhibition of mTOR/p70S6K signaling axis, the dn-AMPKα1 gene was transfected into VSMCs and the cells were then treated with 40 μM telmisartan and 10 ng/mL PDGF for 24 hours. As shown in Fig. 5D, overexpression of dn-AMPKα1 gene was successful as evidenced by the increase in total AMPK expression and the reduction of p-AMPK-Thr172 and p-ACC-Ser79 levels in dn-AMPKα1 gene-transfected cells. Telmisartan inhibition of p-mTOR-Ser2448 and p-p70S6K-Thr389 was significantly reversed in dn-AMPKα1 gene-transfected VSMCs (Fig. 5E), indicating that AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway by telmisartan also occurs in a PDGF-stimulated in vitro atherosclerosis model. Finally, ectopic expression of the dn-AMPKα1 construct significantly attenuated telmisartan-inhibited VSMC proliferation (Fig. 5F). These results suggest that telmisartan inhibits rat VSMC proliferation in PDGF-stimulated cells as well as in PDGF-unstimulated VSMCs via AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway.
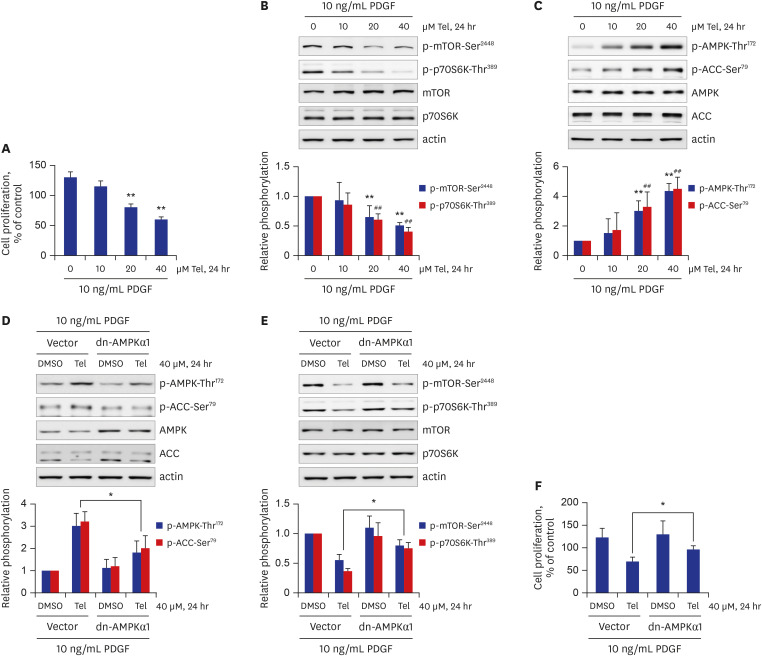 | Fig. 5Telmisartan inhibits PDGF-stimulated VSMC proliferation through AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway. (A) VSMCs were treated with various doses of telmisartan (0, 10, 20, or 40 μM) in the presence of 10 ng/mL PDGF for 24 hours, and subsequent cell proliferation was measured by MTT assay as described in the METHODS. (B, C) VSMCs were treated as described above, and the p-mTOR-Ser2448, p-p70S6K-Thr389, p-AMPK-Thr172, and p-ACC-Ser79 levels were determined using western blot analyses as described in Fig. 1B. (D, E) VSMCs were transfected with rat dn-AMPKα1 (D157A) gene or empty vector, and then treated with 40 µM telmisartan or vehicle (DMSO) in the presence of 10 ng/mL PDGF for 24 hours. The p-AMPK-Thr172, p-ACC-Ser79, p-mTOR-Ser2448, and p-p70S6K-Thr389 levels were detected using western blot analyses as described in Fig. 1B. (F) Rat dn-AMPKα1 (D157A) gene- or empty vector-transfected VSMCs were treated with 40 µM telmisartan or vehicle (DMSO) in the presence of 10 ng/mL PDGF for 24 hours, and subsequent cell proliferation was assessed by MTT assay as described in the METHODS. All experiments were independently performed at least four times, and the blots shown are representative of at least four experiments (n = 4). Bar graphs depict mean fold alterations above/below the control levels (± standard deviation).Differences were considered statistically significant at *P < 0.05, **P < 0.01, and ##P < 0.01.
|
Go to :
DISCUSSION
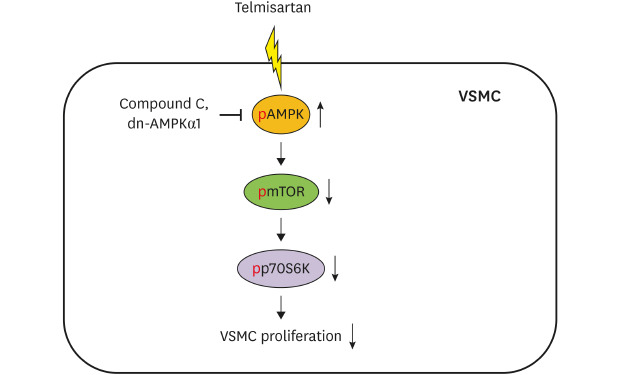
Dysfunction of VSMCs has been reported to contribute to the development of various vascular diseases, including atherosclerosis.13 Excessive and aberrant VSMC proliferation in vasculature is considered a major pathophysiological factor in the development of atherosclerosis and restenosis after angioplasty.614 In the early development of atherosclerotic lesions, VSMCs are reported to produce a variety of inflammatory mediators including TNFα, monocyte chemoattractant protein-1, and vascular cell adhesion molecule-1, as well as the synthesis of extracellular matrix proteins, which ultimately leads to the atheroma formation.1516 Additionally, abnormal VSMC proliferation is reported to be associated with the pathophysiology of in-stent restenosis. Indeed, many patients undergo restenosis due to aberrant VSMC proliferation after balloon angioplasty.17 Therefore, it is likely that attenuation of abnormal VSMC proliferation may be useful in the prevention and treatment of atherosclerosis and restenosis. In this regard, one notable finding in the current study is that telmisartan, through AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway, inhibited VSMC proliferation in both basal and PDGF-stimulated VSMCs (Fig. 6). Based on previous reports and our current results, telmisartan is likely to be useful in the treatment of atherosclerosis and restenosis, as well as in the management of hypertension.
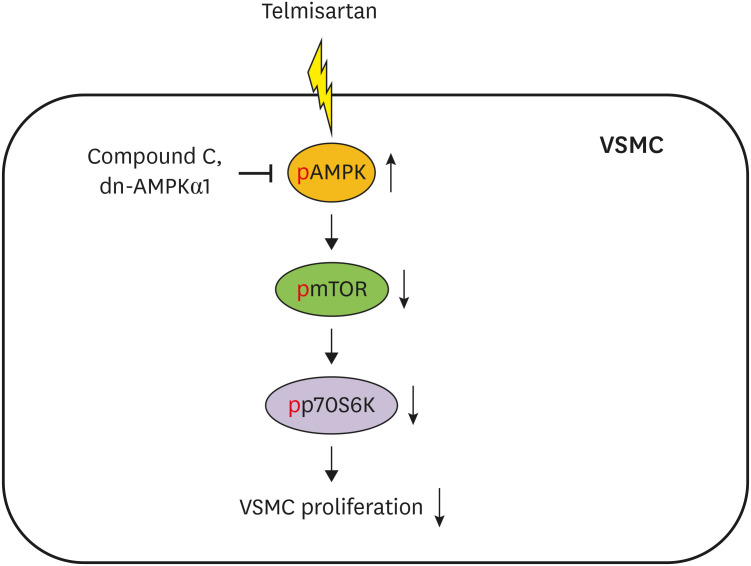 | Fig. 6Schematic illustration of telmisartan inhibition of VSMC proliferation. Telmisartan, not losartan or fimasartan, increases p-AMPK-Thr172 in a PPARγ-independent manner. Increased AMPK activity downregulates p-mTOR-Ser2448 and p-p70S6K-Thr389 levels. Finally, the telmisartan-elevated AMPK activity inhibits VSMC proliferation by decreasing mTOR/p70S6K signaling axis.
|
The mTOR/p70S6K signaling pathway promotes cell proliferation in diverse cell types, including gastric cancer cells, myelogenous leukemia K562 cells, and VSMCs.181920 In particular, phosphorylations of mTOR at Ser2448 and p70S6K at Thr389 are essential for their activation and consequent cell proliferation.21 Furthermore, AMPK activation by 5-aminoimidazole-4-carboxamide ribonucleotide was reported to inhibit the mTOR/p70S6K signaling pathway in human umbilical vein endothelial cells (HUVECs) and VSMCs.2223 Most recently, it was reported that valsartan increased p-AMPK-Thr172 level and decreased p-mTOR-Ser2448 level in rat VSMCs24; however, there were somewhat inconsistent time-course results between AMPK activation and mTOR inhibition, i.e., the decreases in p-mTOR-Ser2448 occurred before the increase in p-AMPK-Thr172.24 Unlike the recently reported data, in the current study, we observed that telmisartan increased AMPK activity first (at 6 hours of treatment) (Fig. 2B) with the mTOR/p70S6K inhibition following at 12 hours of treatment (Fig. 1C). Furthermore, the AMPK inhibition of the mTOR/p70S6K signaling axis was almost completely reversed in the dn-AMPKα1-transfected VSMCs (Fig. 2E). Therefore, it is reasonable to accept that AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway is responsible for telmisartan-inhibited VSMC proliferation (Fig. 6). The inhibitory effect of telmisartan on cell proliferation has also been observed in angiotensin II-stimulated CHO-K1 cells that lack angiotensin II type 1 receptor,6 further supporting our current results showing that the inhibitory effect of telmisartan on cell proliferation is through AMPK-mediated inhibition of the mTOR/p70S6K signaling pathway, which is independent of an angiotensin II type 1 receptor blockade.
Telmisartan, in addition to its customary blood-pressure-lowering effect due to blocking of the body’s RAAS, is reported to have effects that are distinct from other ARBs, such as the capacity to reduce inflammation and improve endothelial dysfunction.25 At present, we cannot fully explain why telmisartan exhibits its distinctive effects other than its common blood-pressure-lowering effects. Based on structural chemistry observations, most ARBs commonly have biphenyl-tetrazole and imidazole groups, whereas telmisartan has a carboxyl group substitute for the common tetrazole group linked to the biphenyl moiety and two benzimidazole groups that are linked tandemly.26 These distinctive structural properties of telmisartan may underlie its ancillary effects. Additionally, in the current study, of the ARBs tested (telmisartan, losartan, and fimasartan), only telmisartan inhibited VSMC proliferation via AMPK-mediated inhibition of the mTOR/p70S6K signaling axis (Fig. 3). Based on these reports and our findings, we speculate that the peculiar proliferation inhibitory effects of telmisartan are associated with its structural differences from other ARBs. It is thought that the second benzimidazole moiety linked to the central one may have a particularly important role in mediating telmisartan's effects.
Telmisartan has been reported to modestly decrease the risk of the composite outcome of cardiovascular death, myocardial infarction, or stroke in the large-scale TRANSCEND trial.27 Furthermore, other large-scale clinical trials, PRoFESS and ONTARGET, have shown potential benefits of telmisartan in reducing secondary strokes.28 Through a combined analysis of PRoFESS and TRANSCEND, the incidence of the composite of stroke, myocardial infarction, or vascular death has been reported to be 12.8% in the telmisartan-administered group, whereas it is 13.8% in the placebo group (P = 0.013).28 However, so far, underlying molecular mechanisms for the beneficial effects of telmisartan on reduction in these vascular complications have not been fully understood. In this regard, our results clearly showed that activation of AMPK by telmisartan inhibited basal and PDGF-stimulated VSMC proliferation via repressing the mTOR/p70S6K signaling axis, which suggests that telmisartan may be able to ameliorate arterial narrowing in pathophysiological circumstances and ultimately reduce various vascular diseases including myocardial infarction and stroke.
The PPARγ protein is one of many ligand-activated nuclear transcription factors and is widely expressed in various cells and tissues,29 including adipose tissue in which it has important roles in the regulation of insulin sensitivity and fat cell differentiation.30 Previously, it has been revealed that telmisartan can stimulate PPARγ transcriptional activity to approximately 30% of the maximal activity level obtained via rosiglitazone, a full PPARγ agonist; therefore, telmisartan was believed to act as a partial PPARγ agonist.1 In this respect, administration of telmisartan in mice has significantly increased serum adiponectin levels and insulin-stimulated glucose use for lipogenesis in mouse adipose tissues.31 Interestingly, these beneficial effects of telmisartan on glucose metabolism are blocked in mice where PPARγ expression is knockout in adipose tissues but not in skeletal muscles,31 indicating a tissue specific role of PPARγ activation by telmisartan in adipose tissues. Furthermore, PPARγ expression levels in human subjects and adult male rats have been reported to be highly variable between several tissues; the most abundant PPARγ expression levels were observed in adipose tissue, large intestine, adrenal gland, and spleen, while PPARγ expression was rarely detectable in skeletal muscles and heart.2932 These differences in PPARγ tissue distribution seem to cause different physiological outcomes of telmisartan in each tissue.
Treatment with telmisartan in a hypertensive chronic kidney disease model rat has improved endothelial function and reduced vascular inflammation in a PPARγ-dependent manner.33 Besides PPARγ activation, telmisartan has also shown its inhibitory effect on cell proliferation in various types of cells, including HUVECs, human aortic smooth muscle cells, and human coronary artery smooth muscle cells, through its ability to inhibit Akt in a PPARγ-independent manner.6 Consistent with these results, we also observed that GW9662, a specific and irreversible PPARγ antagonist, did not affect telmisartan-inhibited VSMC proliferation or telmisartan-decreased AMPK-mediated inhibition of the mTOR/p70S6K signaling axis (Fig. 4). In supporting our results, telmisartan has been previously reported to activate AMPK in 3T3-L1 cells, irrespective of PPARγ activity.34 Based on these previous reports and our current results, it is likely that telmisartan exerts inhibitory effects on VSMC proliferation through a PPARγ-independent pathway. However, further studies are needed to clarify the detailed molecular mechanism by which telmisartan activates AMPK within a PPARγ-independent pathway.
In conclusion, the results in the current study demonstrate that telmisartan-activated AMPK inhibits basal and PDGF-stimulated VSMC proliferation, at least in part, by downregulating the mTOR/p70S6K signaling axis in a PPARγ-independent manner. Elucidation of the detailed mechanism responsible for the inhibitory effects of telmisartan on VSMC proliferation may provide scientific evidence for the usefulness of telmisartan in preventing and treating atherosclerosis and post-intervention restenosis.
Go to :
 XML Download
XML Download