

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Fetal oligohydramnios is defined as an amniotic fluid index (AFI) below 10 percentile corresponding to the gestational age (GA). Severe fetal abnormalities, preterm pre-labor rupture of membranes, and impaired placental function are common causes.1 Severe oligohydramnios, commonly known as AFI less than 5 cm, has a poor prognosis, leading to high mortality after birth. This is due to respiratory failure from pulmonary hypoplasia, especially in the settings of renal dysfunction.12 Renal causes of oligohydramnios include posterior urethral valve, bilateral renal dysplasia, autosomal recessive polycystic kidney disease (ARPKD), and tubular dysgenesis.2
Renal tubular dysgenesis (RTD), is a fatal disorder characterized by faulty developmental of proximal tubules caused by impairment of the renin-angiotensin system (RAS) as a result of mutations of relevant genes (AGT, REN, ACE, and AGTR1) or pharmacological blockade of RAS during early embryogenesis.34567 RAS plays important roles in regulation of blood pressure and development such as ureteric bud branching.38 Therefore, RTD shows deranged renal tubular development, as well as flat-bone ossification because of hypoxemia resulting from hypotension.9 After birth, the patient suffers from persistent hypotension and anuria, which are unresponsive to treatments with volume expanders or high dose catecholamine.10
Here we present a case of RTD in a preterm baby from a pregnancy complicated by severe oligohydramnios from GA 28 weeks. After birth, the patient suffered from persistent anuria and hypotension, and although suspicion for ARPKD was high, identification of ACE gene mutation confirmed the diagnosis of RTD.
Go to : 
CASE DESCRIPTION
A boy, weighed 2,010 g, was born prematurely at GA 33+1 weeks by emergency cesarean delivery due to preterm labor and umbilical cord prolapse in November 2017. Pregnancy was complicated by severe oligohydramnios from GA 28+4 weeks with an AFI close to zero. Fetal kidneys were slightly enlarged (length of 4.2 cm) at GA 29+1 weeks. The mother denied taking any medication during pregnancy and she had no intervention to correct her oligohydramnios.
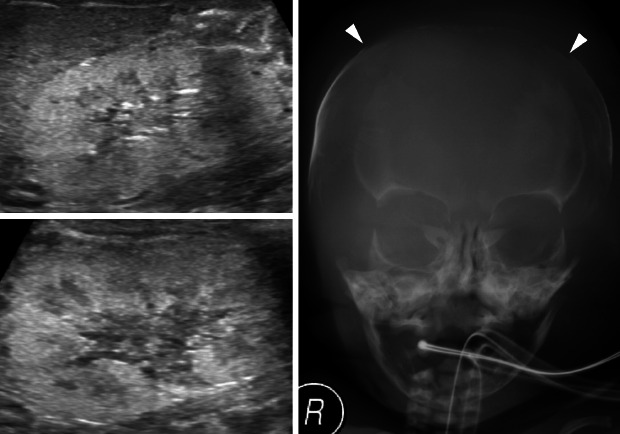
After birth, his heart rate was less than 60 per minutes and no cry was noted. His Apgar score was 1, 5, and 6 at 1, 5, and 10 minutes after birth, respectively. Resuscitation, including chest compression and intubation, was applied upon delivery. Oxygen saturation remained between 70% to 80%, despite surfactant administration, high-frequency oscillation ventilation or nitro oxide and treprostinil administration. Initial cord blood gas analysis showed metabolic and respiratory acidosis with a pH of 6.97, pCO2 104 mmHg, HCO3- 23.9 mmol/L. Initial laboratory findings from cord blood showed urea nitrogen of 5 mg/dL and creatinine 0.34 mg/dL. He was hypotensive; thus, inotropes (dopamine, dobutamine, epinephrine, and vasopressin) and stress dose hydrocortisone for adrenal insufficiency were initiated. However, twenty-four hours after birth, he was noted to have persistent hypotension and remained anuric. His initial infantogram showed a narrow thoracic cage (Fig. 1A). Suspicious of RTD because of anuria and persistent hypotension, skull X-ray was performed and it revealed large fontanelles and wide sutures, suggestive of ossification delay (Fig. 1B). Abdominal sonography on the second day after birth revealed diffuse globular enlargement of both kidneys (right kidney 4.6 cm and left kidney 4.7 cm) with increased cortical parenchymal echogenicity (Fig. 2A). The bladder was collapsed. Abdominal sonography on the third day after birth revealed new bilateral small perinephric fluid collections and multiple dot-like or linear echogenic spots in the right kidney (Fig. 2B). Also, thrombi in inferior vena cava were shown in ultrasonography (Fig. 2C). Heparin was initiated; however, two days later, expansion of the thrombi was noted now involving both renal veins.
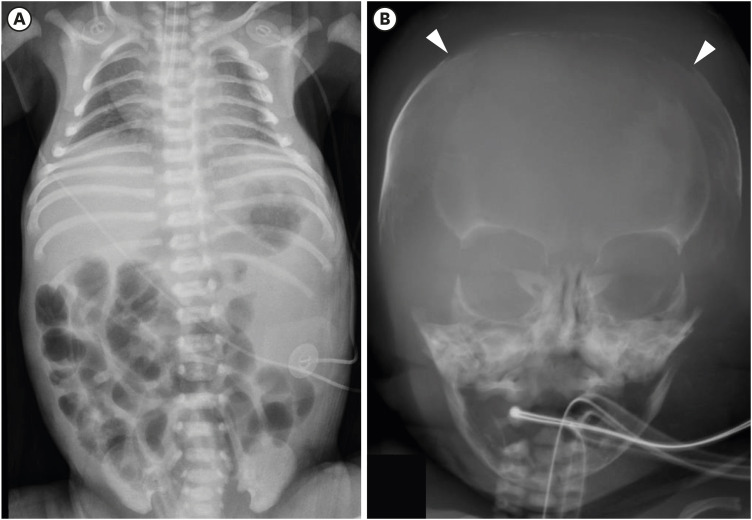 | Fig. 1The simple X-ray images of the patient showing characteristics of RTD. (A) Initial infantogram showing narrowing thoracic cage with the intubated state. (B) Large fontanelle and wide suture suggesting ossification delay.
|
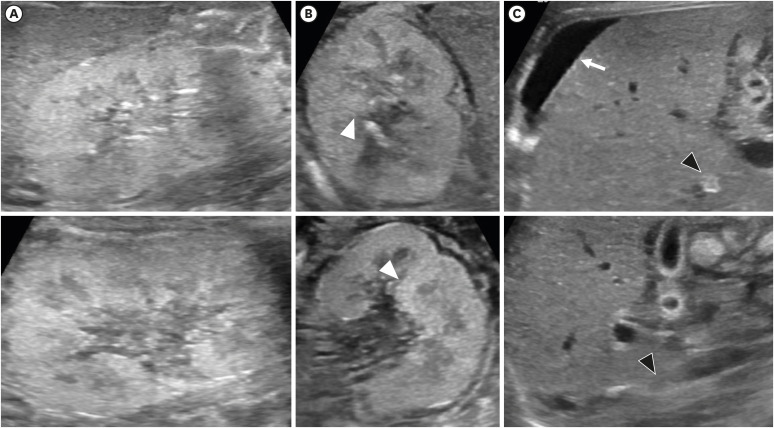 | Fig. 2Kidney sonography of the patient (the upper row: right kidney, the lower row: left kidney). (A) At the next day after birth, sonography showed diffuse globular enlargement of both kidneys (right kidney 4.6 cm and left kidney 4.7 cm) with increased cortical parenchymal echogenicity. (B) Third day of birth, newly-occurred multiple dot-like or linear echogenic spots (white arrow heads) were found in the both kidney. (C) Also, new bilateral small perinephric fluid collection (white arrow) and segmental obliterations of IVC in intrahepatic and suprarenal segment were detected, suggesting thrombi in the IVC (black arrow heads).IVC = inferior vena cava.
|
Serum basal renin was 120.0 ng/mL/hr and 22.09 ng/mL/hr at 4 days and 10 days after birth, respectively. Basal aldosterone was 17.09 ng/dL and 24.5 ng/dL at 4 days and 10 days after birth, respectively. Also, on the 10th day, serum angiotensin-converting enzyme (ACE) was not detectable (below 5.0 U/mL) and angiotensin II (ANG II) was 312 pg/mL. Serum creatinine increased to 3.09 mg/dL at the 10th day. Normal reference intervals of serum hormones are described in Table 1.11 Genetic analyses revealed 2 frame-shifting duplications in the ACE gene (REFSEQ: NM_000789.3): c.1454dupC in exon 9 (p.Ser486Phefs) and c.2141dupA in exon 14 (p.Asn714Lysfs) (Fig. 3). Both variations have not been previously reported. Genetic testing of the parents was not available.
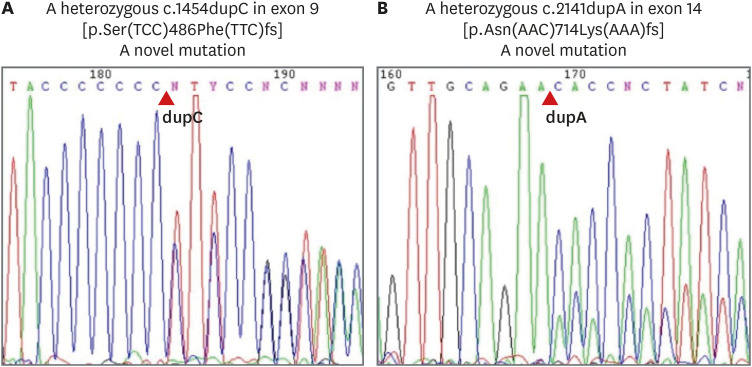 | Fig. 3The result of genetic analysis showing variations in ACE gene. (A) A heterozygous c.1454dupC in exon 9 (p.Ser486Phefs). (B) A heterozygous c.2141dupA in exon 14 (p.Asn714Lysfs).
|
Table 1
Reference interval of serum hormone at age of 1 week to 3 monthsa
| Variables | Median | Range |
|---|---|---|
| Basal renin, ng/mL/hrb,c | 12.0 | 7.1–23.8 |
| Aldosterone, ng/dLb,d | 62 | 30–201 |
| ACE, U/mLe | 21.6 | 12.1–32.9 |
| ANG II, pg/mLf | 58 | 30–117 |
ACE = angiotensin-converting enzyme, ANG II = angiotensin II.
aAll blood sample was taken between 8:30 and 9:00 in the morning with supine posture and fasting since midnight (Infants fasted 3 hours before blood sample) in healthy patients and put into ice-cooled tubes. Plasma was immediately separated and frozen at −20°C until assay; bThe tube had 10 mg potassium EDTA; cMeasured by Clinical Assays, the commercial kit; dMeasured by direct radioimmunoassay; eThe tube had heparin. The sample was measured using hiuppuryl-L-histidyl-L-leucine as a substrate and spectrophotometrically at 228 nm; f0.125 M EDTA and 0.025 M O-phenanthroline in distilled water as inhibitors. The sample was measured by direct radioimmunoassay in unextracted plasma.
![]()
Despite the continuous infusion of inotropes and antibiotics, his hypotension persisted and he remained anuric. Peritoneal dialysis was started from the second day after birth for suspicious ARPKD but with frequent interruptions. Hyponatremia as low as 123 mmol/L on day three, improved, without reflex hyperkalemia, after 0.05 mg fludrocortisone was initiated on day six. The changes of blood pressure with interventions were shown in Fig. 4. On the 23rd day after birth, desaturation and bradycardia rapidly worsened, resulting in cardiac arrest. He then expired without further resuscitation following the wishes of his parents.
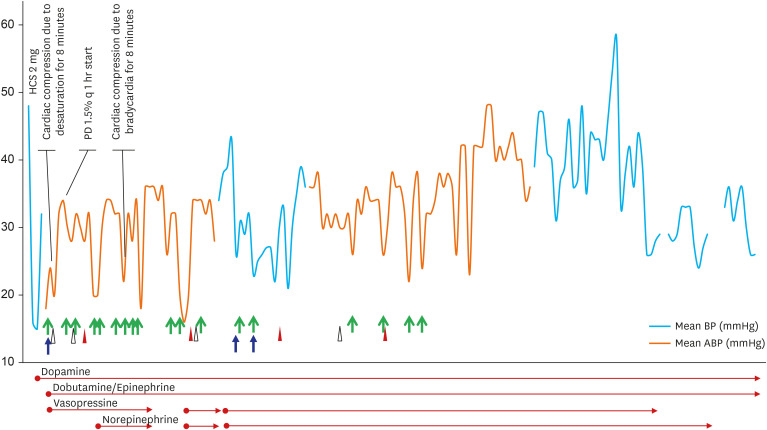 | Fig. 4The changes of patient's mean blood pressure associated with interventions such as inotropes, volume repletion or PD. 5% albumin (green arrows), normal saline (blue arrows), and blood (red arrow heads for red blood cell and white arrow heads for fresh frozen plasma) was used for volume repletion.HCS = hydrocortisone, PD = peritoneal dialysis.
|
Ethics statement
This case was reviewed and approved by the Institutional Review Board (IRB) of Seoul National University Hospital (IRB No. 0812-002-264). Requirement for informed consent was waived after review of IRB because it was practically impossible and this study was of retrospective design.
Go to :
DISCUSSION
RTD is a fatal and rare disease with an unknown incidence.49 RTD is caused by loss of function of the renal proximal tubule and chronic hypoperfusion of the fetal kidney, similar to what occurs in twin-twin transfusion syndrome, renal artery stenosis, or fetal exposure to RAS blockers.310 RTD is usually inherited in an autosomal recessive pattern.3 Variations result in the absence or decrease of ANG II production or function, leading to refractory hypotension.410 Thrombosis and skull ossification defect, as seen in our case, can be explained by this persistent fetal-neonatal hypotension.412
In our patient, RTD was induced by two novel 1-base pair duplications confirmed in genetic analysis. Both variations result in frame-shifting with functional impairment (functional impact scores predicted by MutationAssessor [http://mutationassessor.org/] were 3.015 [c.1454dupC] and 2.375 [c.2141dupA]). Although there is a limitation interpretation because genetic testing in the parents were not performed, both variations were considered to be ‘pathogenic variations’ according to the guidelines13 developed by the American College of Medical Genetics and Genomics (1 very strong [PVS1], 1 moderate [PM2], and 1 supporting [PP4] evidence).
Laboratory findings also supported the diagnosis of genetic RTD in this case; In RTD with RAS mutations, except REN, the renin expression is usually increased as a result of negative feedback on the RAS pathway.3412 Since ACE was mutated in our patient, serum ACE and aldosterone levels were low,14 but, serum ANG II was increased above the normal range of age. Considering that serum ANG II increases after birth in preterm babies,15 it can explain the increase of ANG II in our patient.
Initially, we suspected ARPKD, since oligohydramnios was severe along with anuria. However, ultrasonography findings were not typical of ARPKD; kidneys in ARPKD are highly echogenic and enlarged.16 On the other hand, kidneys affected by RTD have normal or slightly enlarged sizes with or without normal echogenicity and loss of corticomedullary differentiation as seen in our case. Because these findings are not specific, it is difficult to diagnose RTD by fetal kidney sonography only, although the sensitivity of prenatal ultrasonography screening for renal malformation is now up to 80%.17 In addition to renal ultrasonography, a typical pre-natal finding of RTD is severe oligohydramnios, which manifests around GA 20 weeks.9 In our patient, oligohydramnios was first detected at GA 24 weeks and severe oligohydramnios at GA 28 weeks. Also, antenatal renal ultrasonographic findings were compatible with RTD.
Hypotension is the major challenge during the first days of life in patients with RTD. While vasopressin was reported as helpful to remedy hypotension in survived cases,1418 it was not effective in our case. Volume repletion using volume expanders such as fresh frozen plasma, which reversed hypotension in a few cases,414 was not effective either. There are reports that fludrocortisone can be used for electrolyte stabilization and can significantly improve clinical outcomes,1418 but in our patient, fludrocortisone was helpful only in electrolyte stabilization.
Despite rigorous management, our patient did not survive. To date, 14 patients with genetically confirmed RTD have survived according to the literature. Almost all the reported patients, similar to our patient, needed renal replacement therapy, and all the survived cases except one had chronic kidney disease.12141920 In Korea, only one case was reported until now, and that patient survived with chronic kidney disease stage 2.19
Interestingly, prognosis, severity, or the phenotype of RTD is not correlated with locus heterogeneity or differences in the mutated genes.412 Rather than the specific type of variations or causative gene, response to supportive care for respiratory difficulty and hypotension is more important for the neonatal survival;10 rapid recovery of diuresis is considered a favorable prognostic factor.101419
As presented in this report, despite its rarity, suspicion for RTD should be high in preterm neonates with refractory hypotension whose gestational course was complicated by severe oligohydramnios.12 Faulty ossification of skull bones with wide sutures and high renin levels support the diagnosis of RTD. For genetic analysis, molecular identification of pathogenic variations in RAS-associated genes can confirm the disease, opening the possibility for genetic counseling.12
Go to :
 XML Download
XML Download