

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Congenital or hereditary hemolytic anemia (HHA) occurs when red blood cells (RBC) have inherited genetic mutations causing congenital defects in RBC membrane proteins, globin chains, or RBC enzymes.1 HHA is a rare but very heterogeneous group of RBC disorders generally classified as RBC membranopathies, hemoglobinopathies, and RBC enzymopathies, depending on the etiology.1 As HHA is a lifelong chronic RBC disorder, long term regular examinations and treatment are needed to reduce complications due to the disease itself and repeated blood transfusions.2 Hereditary spherocytosis (HS), a typical type of RBC membranopathy, is the most common HHA in Korea.3 Hemoglobinopathies cause significant health problems in over 70% of 229 countries4; however, they have been rarely diagnosed in Korea.3 RBC enzymopathies are suspected when a child shows non-immune hemolytic anemia with normal RBC morphology and a normal RBC index, after differentiating RBC membranopathy and hemoglobinopathy, and are also very rarely reported in Korea.3 Recently, accurate diagnoses of RBC membranopathies and enzymopathies are possible due to improved membrane protein or enzyme analysis methods such as liquid chromatography-tandem mass spectrometry (LC-MS/MS),5 ultra-performance liquid chromatography-tandem mass spectrometry (UPLC-MS/MS),5 and high performance liquid chromatography techniques,6 and more accurate screening methods including flow cytometric osmotic fragility (OF) test789 and eosin-5′-maleimide (EMA) binding test.7
In the current era, the epidemiology of hemoglobinopathies in the European population have changed due to the migration of various populations from several parts of the world to European countries.101112 In Korea, there has been an increase in female who immigrated from South-East Asia through international marriages, particularly in the rural areas.1314 We considered that the change in domestic situations may have affected the overall Korean HHA epidemiology. To perform nationwide epidemiologic studies of rare HHA, standard diagnostic guidelines and central laboratories are needed. Therefore, the Korean Society of Hematology (KSH) has been operating the HHA Working Party (WP) since 2005; the name changed to “RBC Disorder WP” in November of 2016.351516 The RBC Disorder WP developed the Korean standard operating procedure (SOP) for diagnosis of HHA in 2007 and continuously updated the SOP to ensure accurate and nationwide standardized diagnostic methods and reporting of laboratory tests.5 In this epidemiologic study of pediatric HHA in Korea, we collected data from a newly diagnosed cohort of children with HHA according to the Korean SOP for HHA, from 2007 to 2016, and compared this to data collected from a previous cohort, from 1997 to 2006, to assess whether the epidemiology of HHA in Korea has changed.
The aim of this study was to conduct a nationwide multicenter epidemiologic study of HHA in Korea every 10 years, to compare this to the previous 10-year-cohorts through KSH, to evaluate and upgrade the Korean SOP for HHA, to increase the awareness among clinicians of the possibility of HHA in Korean anemia patients, and to determine whether new diagnostic methods should be developed for making an accurate diagnosis of HHA.
METHODS
Diagnosis of HHA
The members of KSH diagnose HHA according to Korean SOP of HHA, and this SOP is shown in Fig. 1. To determine defects in the RBC membrane proteins in HHA, mass spectrometry was performed in a central laboratory.515 The RBC Disorder WP applied LC-MS/MS for analyzing RBC membrane proteins in the central laboratory of Korea since March 2013.5 LC-MS/MS is more accurate and less time consuming for protein quantification than sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) because, in LC-MS/MS, each protein is measured separately without being affected by band 3 deficiency.5 Flow cytometric OF and EMA binding testing were registered in the New Health Technology Assessment of Korea in November 2014 and have been used since then.79 Globin gene mutations were diagnosed by direct sequencing and deletion analysis (multiple ligation-dependent probe amplification, MLPA) of the beta-globin and then alpha-globin gene. RBC enzymes were analyzed by multiplex RBC enzyme analysis using the UPLC-MS/MS in the central laboratory of Korea since March 2013.5 The 10 RBC enzymes assessed were adenosine deaminase, adenylate kinase, glucose-6-phosphate dehydrogenase, glucose phosphate isomerase, hexokinase, phosphofructokinase, phosphoglycerate kinase, pyrimidine 5′-nucleotidase, pyruvate kinase, and triosephosphate isomerase.5
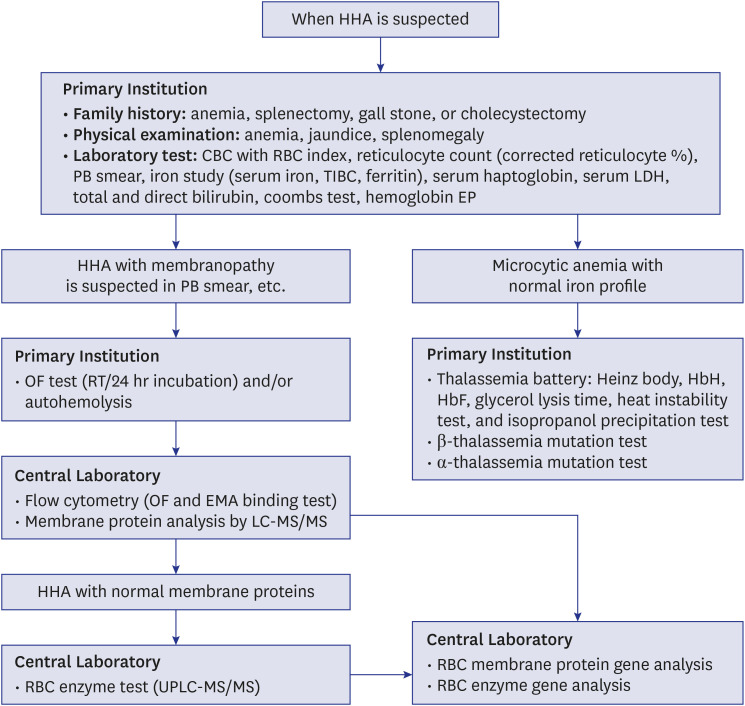
Fig. 1
Standard operating procedure for the diagnosis of HHA by the RBC Disorder Working Party of the Korean Society of Hematology.515
HHA = hereditary hemolytic anemia, CBC = complete blood count, RBC = red blood cell, PB = peripheral blood, TIBC = total iron binding capacity, LDH = lactate dehydrogenase, EP = electrophoresis, OF = osmotic fragility, RT = room temperature, EMA = eosin 5-maleimide, LC-MS/MS = liquid chromatography-tandem mass spectrometry, UPLC-MS/MS = ultra-performance liquid chromatography-tandem mass spectrometry.
![]()
Subject data collection
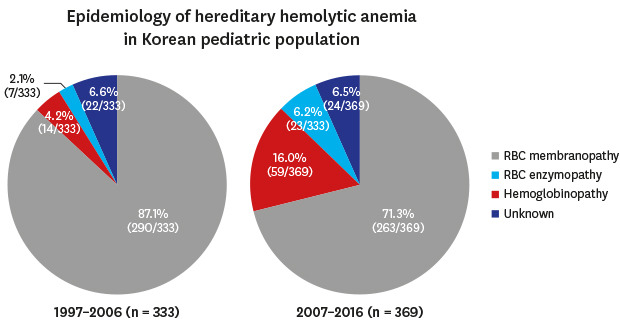
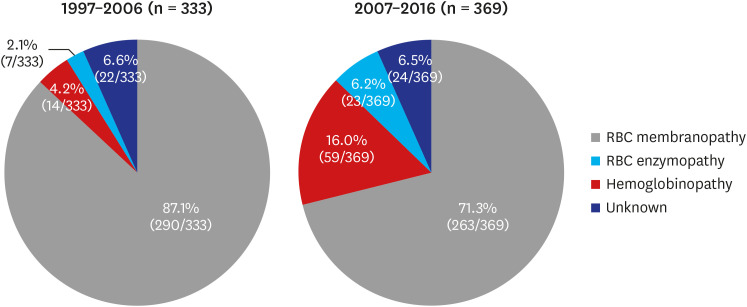
The study was performed as part of an official research policy to identify the status of rare RBC disorders in Korea in a 10-year cycle, through the KSH. The 10-year epidemiology of Korean HHA was previously investigated from 1997 to 2006.3 In the previous 10-year study (1997–2006), 431 newly diagnosed HHA patients’ data were collected and analyzed, and among them, 333 patients were children. Among the 333 newly diagnosed pediatric HHA patients, RBC membranopathies accounted for 290 (87.1%), hemoglobinopathies for 14 (4.2%), RBC enzymopathies for 7 (2.1%), and unknown etiology of non-immune HA for 22 (6.6%). The most common HHAs were RBC membranopathies, accounting for 87.1%.3 On the other hand, hemoglobinopathies (4.2%) and RBC enzymopathies (2.1%) were very rare.3
In this present 10-year study (2007–2016), patient information, clinical manifestations, and laboratory findings of Korean pediatric HHA patients newly diagnosed from January 2007 to December 2016 were retrospectively collected using a questionnaire-based survey of Korean pediatric hematologists. The survey questionnaire was sent to all KSH members in the field of pediatric hematology who were working at secondary or tertiary medical centers in Korea. The questionnaire was prepared in Excel by the HHA WP and collected through formal e-mail by the KSH. The questionnaire included the following items: the name of the medical center, initials of the patients' names, the patients' sex, the patients' birth dates, date of diagnosis of HHA, clinical diagnosis, family history of the same disease (including parents, siblings, and other family members), ethnicity of parents, the patients' symptoms and signs at the time of diagnosis of HHA, Hb, mean corpuscular volume (MCV), mean corpuscular hemoglobin (MCH), mean corpuscular hemoglobin concentration (MCHC), corrected reticulocyte, total bilirubin, direct bilirubin, LDH, haptoglobin, and ferritin values, the results of a direct and an indirect Coombs test, the results of an OF test at room temperature and 24 hour incubation, the result of autohemolysis tests, the results of RBC membrane protein analysis, the existence of Hb H inclusion, the existence of Heinz bodies, the ratio of Hb A/A2, the percent of Hb F, isopropanol instability, the genetic mutations found, and RBC enzyme levels.
The patients were diagnosed according to the Korean SOP for HHA.515 The duplicate patients were removed based on the date of birth and the initials of the patient's name. In addition, if the collected data reflected autoimmune hemolytic anemia (AIHA), paroxysmal nocturnal hemoglobinuria (PNH), myelodysplastic syndrome (MDS), or congenital dyserythropoietic anemia (CDA), they were excluded from the study.
Statistical analysis
The variables were described as median values and ranges. To compare the ratio of variables according to the disease categories, linear-by-linear association was evaluated with χ2 tests. To compare the median values of laboratory tests between the disease categories, a Kruskal-Wallis test as well as a Mann-Whitney U test with Bonferroni's correction was employed. P values of < 0.05 were considered statistically significant. Statistical analysis was performed using SPSS ver. 23 (SPSS Inc., Chicago, IL, USA). To determine the number, percentage, and the trend of recent Korean domestic marriages with foreigners, information from the Korean Statistical Information Service (KOSIS, https://kosis.kr) was applied.17
Ethics statement
This multicenter retrospective study was approved by the Institutional Review Board (IRB) of each participating institution (Keimyung University Dongsan Medical Center IRB Approval No. 2017-08-020, Sungkyunkwan University Kangbuk Samsung Hospital IRB Approval No. 2017-10-012-001, and Seoul National University Hospital IRB No. 1911-157-1082), and the requirement for informed consent was waived.
RESULTS
Subject collection
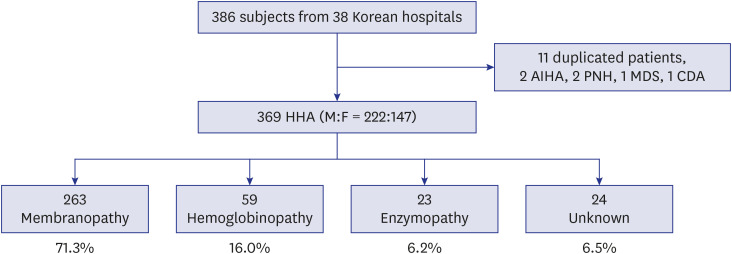
The medical institutions and pediatric hematologists who answered the survey in this present 10-year study (2007–2016) covered 16 of the 17 administrative districts of Korea; 7 Metropolitan Cities (Seoul, Incheon, Daejeon, Daegu, Gwangju, Ulsan, and Busan) and 9 provinces (Gyeonggi, Chungbuk, Chungnam, Jeonbuk, Jeonnam, Gangwon, Gyeongbuk, Gyeongnam, and Jeju Special Self-Governing Province). Since there are no pediatric hematologists in Sejong Special Self-Governing City, this city was omitted from this survey. In the previous 10-year study (1997–2006), the Jeju Special Self-Governing Province was not included from the survey because there were no pediatric hematologists on Jeju island at that time. However, this present 10-year study (2007–2016) was conducted by additionally including the Jeju Special Self-Governing Province.
Consequently, data of 386 participants were collected from 38 hospitals in Korea. Among them, 11 duplicated patients, 2 patients with AIHA, 2 patients with PNH, 1 patient with MDS, and 1 patient with CDA were excluded. Finally, 369 children with newly diagnosed HHA (male:female = 222:147) were analyzed. Among 369 pediatric HHA patients, RBC membranopathies accounted for 263 (71.3%), hemoglobinopathies for 59 (16.0%), RBC enzymopathies for 23 (6.2%), and unknown etiology of non-immune HA for 24 (6.5%). The collection process of HHA data and the distribution of institutions are shown in Fig. 2. The data from this 10-year (2007–2016) cohort were compared with the previous 10-year (1997–2006) cohort data.3 These comparisons showed that the ratio of hemoglobinopathies had significantly increased from 4.2% (14/333) to 16.0% (59/369) (P < 0.001). Moreover, the ratio of RBC enzymopathies had significantly increased from 2.1% (7/333) to 6.2% (23/369) (P = 0.008). On the other hand, the ratio of RBC membranopathies decreased from 87.1% (290/333) to 71.3% (263/369) (P < 0.001), and the proportion of non-immune hemolytic anemia with unknown etiology was similar (P = 1.000). The changes in HHA distribution in Korea during 20 years are shown in Fig. 3.
Clinical manifestations and laboratory data of the three HHA groups
Table 1 shows the 369 HHA patients’ clinical characteristics classified according to etiologies, which include initial symptoms/signs or the main reason for visiting the hospital and conducting the HHA test and the results of laboratory tests by disease group: Hb, MCV, MCH, MCHC, corrected reticulocyte, bilirubin, LDH, and haptoglobin values. Although not statistically significant, the age of subjects with RBC enzymopathies at diagnosis was the lowest (median age, 0.8 years) among the three HHA groups. Skin pallor was most common among RBC enzymopathies, followed by RBC membranopathies and hemoglobinopathies (P = 0.014). Jaundice and splenomegaly were most common among RBC membranopathies, followed by hemoglobinopathies or RBC enzymopathies (P < 0.001 and P = 0.004, respectively). Asymptomatic patients were most common among hemoglobinopathies, followed by RBC enzymopathies or RBC membranopathies (P < 0.001). The median value of Hb was lowest in the RBC enzymopathies (7.5 g/dL) and highest in the hemoglobinopathies (10.7 g/dL). The median values of the RBC profiles including MCV, MCH, and MCHC were all lowest in the hemoglobinopathies. The median value of corrected reticulocytes was lowest in the hemoglobinopathies (1.2%) and highest in the RBC membranopathies (5.5%). The median value of LDH was lowest in the hemoglobinopathies (385 IU/L) and highest in RBC enzymopathies (856 IU/L). The median value of haptoglobin was lowest in the RBC membranopathies (7.8 mg/dL) and highest in the hemoglobinopathies (82 mg/dL).
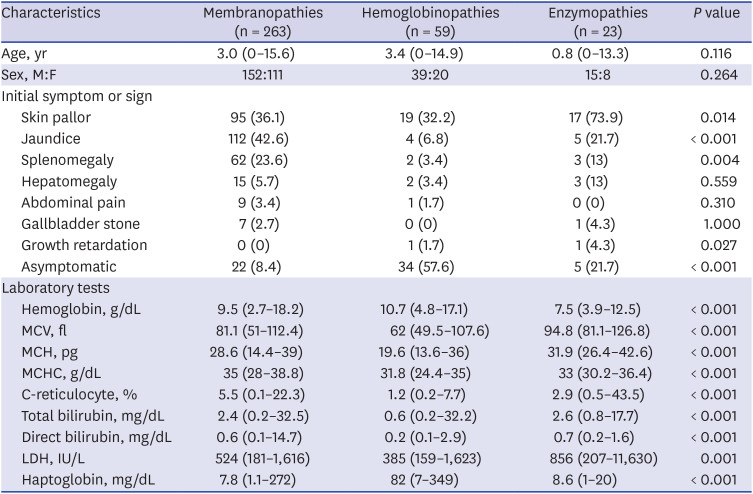
Table 1
Clinical characteristics of children with hereditary hemolytic anemia in Korea
Data are presented as median (range) or number (%).
MCV = mean corpuscular volume, MCH = mean corpuscular hemoglobin, MCHC = mean corpuscular hemoglobin concentration, C-reticulocyte = corrected reticulocyte, LDH = lactate dehydrogenase, M = male, F = female.
![]()
RBC membranopathies
The total number of children with RBC membranopathies was 263 (male:female = 152:111). There were 253 patients with HS and 10 with hereditary elliptocytosis. The median age at diagnosis was 3 years (range, 0–15.6 years). Among the 263 children, 129 patients (49.1%) had a family history of the same disease. Jaundice (112/263, 42.6%) was the most common finding in children with RBC membranopathies, followed by skin pallor (95/263, 36.1%) and splenomegaly (62/263, 23.6%). The median value of Hb was 9.5 g/dL (range, 2.7–18.2 g/dL) and the median value of corrected reticulocytes was 5.5% (range, 0.1%–22.3%). Among 263 children, 23 (8.7%) had corrected reticulocytes < 1% at the time of diagnosis, suggesting that they were diagnosed with RBC membranopathies in a state of aplastic crisis. According to the previously published report about the genetics of Korean HS patients through RBC disorder WP, the most frequent genetic variants in Korean HS were found in SPTB, followed by ANK1, and SLC4A1.15
Hemoglobinopathies
There were 59 subjects (male:female = 39:20) with hemoglobinopathies. The fraction of children who were newly diagnosed with hemoglobinopathies in recent 10 years (2007–2016) was estimated to have increased by nearly 4-fold as compared to that in previous 10 years (1997–2006) (Fig. 3). The median age at diagnosis was 3.4 years (range, 0–14.9 years). Among these 59 patients, 49 children had beta-thalassemia minor and 9 children with alpha-thalassemia minor while 1 child was diagnosed with unstable hemoglobin. Twenty-five of 59 patients with hemoglobinopathies had a family history of the same disease. Among 59 patients, 36 patients (61%) had both parents of Korean ethnicity, but 23 patients (39%) had mothers immigrated from South-East Asia who were from Vietnam (15), Cambodia (4), China (1), Thailand (1), Philippines (1), and Singapore (1). Among the 59 patients, 37 patients were diagnosed with hemoglobinopathies by globin DNA analysis. The thalassemia variants and the ethnicity of the parents are shown in Table 2. The remaining 22 patients were diagnosed with hemoglobinopathies by family history and Hb electrophoresis. Asymptomatic subjects were most common (34/59, 57.6%) among children with hemoglobinopathies, and 32.2% of patients had skin pallor. The median value of Hb was 10.7 g/dL (range, 4.8–17.1 g/dL), and the median value of corrected reticulocytes was 1.2% (range, 0.2%–7.7%).
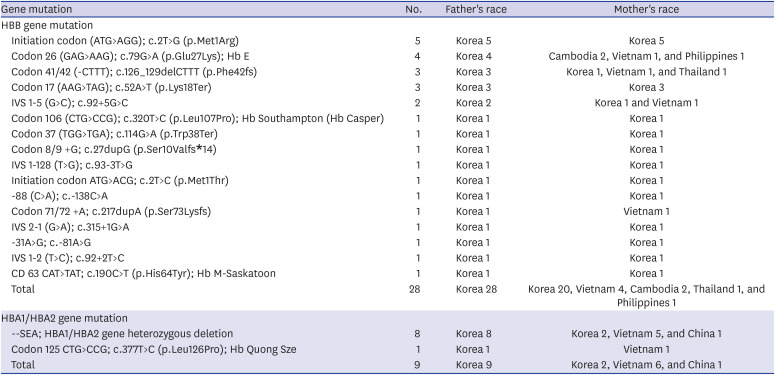
Table 2
Genetic variants and races of the parents of pediatric hemoglobinopathy patients in Korea
![]()
During the study period, the total number of Korean marriages from 2000 to 2016 could be obtained from the Korean Statistical Information Service (Supplementary Table 1 and Supplementary Fig. 1). This data showed that, since 2003, the number of marriages to foreign female (marked in red color) had increased. The number of marriages to foreign female increased sharply from 2003, peaked in 2005, and decreased slightly since then. Considering that the median age at diagnosis of newly diagnosed hemoglobinopathy in this study was 3.4 years, it may be related to the increase in the number of marriages to immigrants.
RBC enzymopathies
The total number of children diagnosed with RBC enzymopathies from 2007 to 2016 was 23 (M:F = 15:8) including 12 patients with pyruvate kinase (PK) deficiencies, 9 patients with glucose-6-phosphate dehydrogenase (G6PD) deficiencies, 1 patient with enolase deficiency, and 1 patient with 2 or more enzyme deficiencies. The proportion of children who had been newly diagnosed with enzymopathies in recent 10 years (2007–2016) was estimated to have increased nearly 3-fold as compared to that in previous 10 years (1997–2006) (Fig. 3). The median age at diagnosis of enzymopathy was 0.8 years (range, 0–13.3 years). Six of the 23 patients with enzymopathies had a family history of the same disorder. Compared with the data of the previous 10 years, PK deficiency had significantly increased. Skin pallor was the most common symptom (17/23, 73.9%) among children with RBC enzymopathies, 21.7% of patients had jaundice, and another 21.7% were asymptomatic. The median value of Hb was 7.5 g/dL (range, 3.9–12.5 g/dL), and the median value of corrected reticulocytes was 2.9% (range, 0.5%–43.5%).
DISCUSSION
In Korea, RBC Disorder WP of KSH established the Korean SOP for HHA in 2007, and continuously updated for nationwide standardization of diagnosis of HHA, a rare hematologic disease. In this study, we conducted the nationwide multicenter epidemiologic study of HHA in Korea diagnosed according to the updated the Korean SOP for HHA with advanced diagnostic techniques. We are performing this nationwide multicenter epidemiologic study of HHA every 10 years to compare with the previous 10-year-cohorts through KSH. The purpose of this study was to evaluate and upgrade the Korean SOP for HHA, to increase the awareness among clinicians of the possibility of HHA in Korean anemia patients, and to determine whether new diagnostic methods are necessary to develop for the accurate diagnosis of HHA.
This study investigated the incidence of newly diagnosed pediatric patients with HHA from 2007–2016 and compared this incidence to that of an HHA cohort from 1997–2006 in Korea. From this study, significant increases in hemoglobinopathies (4.2% to 16.0%) and RBC enzymopathies (2.1% to 6.2%) were observed. RBC membranopathies were found to be the most common HHA (87.1% to 71.3%). The RBC disorder WP of KSH used several improved diagnostic techniques and improved SOP for diagnosing Korean HHA patients. In addition, in the case of membranopathies, a multi-gene targeted sequencing method was applied, and the results were published by Choi et al.,15 on the behalf of the RBC disorder WP, that showed that the most frequent genetic variants in HS were found in SPTB, followed by ANK1 and SLC4A1.15
According to the data regarding the Korean population dynamics trend from KOSIS,17 the number of births and the birth rate in Korea has persistently decreased. The number of births was decreased from 675,394 in 1997 to 406,243 in 2016, and the crude birth rate per 1,000 people also decreased from 14.5 in 1997 to 7.9 in 2016.17 The fertility rate per 1 female of childbearing potential was also decreased from 1.537 in 1997 to 1.172 in 2016.17 Thus, we suggest that the real number of newly diagnosed pediatric HHA cohort have also been decreased according to the trend of Korean marriages and births. For this reason, we believe that the number of RBC membranopathy patients among the total HHAs may have decreased. Nevertheless, it is thought that the proportion of hemoglobinopathy and enzymopathy increased as marriages to foreigners increased, and specialized diagnostic testing skills developed.
In the case of hemoglobinopathies, there have been some single-center reports of hemoglobinopathies due to increasing rates of international marriage in recent years in Korea.1618 Hemoglobinopathies are known to occur in the vicinity of the Mediterranean.4 If people emigrate from their original country to a new country, the genetic disease dynamics of those countries will change. For example, major hemoglobinopathies such as thalassemia syndromes and sickle cell disorders, are rare diseases, but their prevalence is significantly growing in many countries due to mobility and migratory flows.10 Furthermore, a recent observational study in healthy Korean individuals < 30 years old, from 2015 to 2017, showed that the prevalence of thalassemia in young Koreans was increasing, which was associated with an increase in the number of South-East Asian immigrants.19
In our study, subjects with hemoglobinopathies showed the highest Hb, lowest corrected reticulocyte, lowest LDH, and highest haptoglobin values among the three HHA groups. Therefore, Korean patients with hemoglobinopathies may be the least symptomatic. Since the majority of Korean patients with hemoglobinopathies have thalassemia minor, hemolysis among these patients was not clinically significant.
Commonly found variants for Korean β-thalassemia were initiation codon (ATG>AGG); c.2T>G (p.Met1Arg, and codon 17 (AAG>TAG); c.52A>T (p.Lys18Ter), resulting in β0 in 8 patients in this study. The parents of these patients were all Koreans; initiation codon (ATG>AGG) and codon 17 (AAG>TAG) are known to be originally distributed in Korea, China, and Japan.2021 In addition, the variants of β-thalassemia, codon 26 (GAG>AAG); c.79G>A (p.Glu27Lys); Hb E, codon 41/42 (-CTTT); c.126_129delCTTT (p.Phe42fs), IVS 1-5 (G>C); c.92+5G>C, and codon 71/72 +A; c.217dupA (p.Ser73Lysfs) mainly occurred in South-East Asian parents (Vietnam 4, Cambodia 2, Philippines 1, and Thailand 1). By molecular analysis of β-thalassemia in Vietnam over a 15-year period, codon 26 (GAG>AAG) (29.2%) and codon 41/42 (-CTTT) (18.8%) were found to occur frequently.22 In the case of α-thalassemia, the common variant was a SEA deletion, resulting in an α0-thalassemia allele phenotype, mainly from a Vietnamese mother. In a study of the spectrum of α-globin deletions in Vietnam, SEA deletion was the most common deletion mutation (87.35%).23 Among the 37 patients who have been genetically diagnosed as hemoglobinopathies, there were 23 patients with both parents of Korean ethnicity, showing an increment in incidence when compared to the previous 10-year study (Table 2). Therefore, it is believed that increased awareness of clinicians about the possibility of hemoglobinopathy as the cause of HHA in Korean anemia patients has also been an important contributing factor in increasing the number of patients with hemoglobinopathies in the 2007–2016 study.
β-thalassemia results in markedly aberrant Hb electrophoresis. Therefore, several Korean β-thalassemia patients were diagnosed based on family history and Hb electrophoresis, without genetic testing. However, since HBB gene sequencing is not very expensive in Korea, genetic tests were often performed when medical staff suspected β-thalassemia. In contrast, in the case of α-thalassemia, Hb electrophoresis test results are normal, and MLPA of HBA1/HBA2 can diagnose the most common deletions. In Korea, however, MLPA is not covered by the National Health Insurance System and is therefore very expensive. Thus, in Korea, the diagnosis of α-thalassemia is considered to be underestimated.
RBC enzymopathies generally present with normocytic normochromic hemolytic anemia, with normal morphology of RBCs in the peripheral blood.24 Thus, if a patient shows non-immune hemolytic anemia without the characteristic findings of RBC membranopathies or hemoglobinopathies, enzymopathies are clinically suspected. The diagnosis of enzymopathies is based on the detection of reduced specific enzyme activity and molecular characterization of the defect at DNA level.2425 In our study, subjects with enzymopathies were very young at diagnosis (median age, 0.8 years). Furthermore, the Hb and haptoglobin levels were relatively low, and LDH levels were high. Considering these characteristics, patients with enzymopathies have a distinct hemolytic phenotype at a very young age.
G6PD is known to be implicated in the most common type of RBC enzymopathy, with X-linked inheritance, worldwide.126 G6PD-deficiency is common throughout the Mediterranean, Africa, and the Middle East, affecting more than 400 million people worldwide.2627 The prevalence of G6PD-deficiency correlates with the geographical distribution of malaria, leading to the theory that carriers of G6PD-deficiency may have partial protection against malaria infection.2627 G6PD plays an essential role in the production of nicotinamide adenine dinucleotide phosphate, which plays an important role in preventing oxidative damage to the RBC.26 Although the exact prevalence of the G6PD-deficiency in Korea is not yet known, the interest in G6PD-deficiency and diagnosis has been increasing recently.28 PK-deficiency is considered the second most common RBC enzymopathy worldwide,129 but we found it to be more frequent than G6PD-deficiency in the present study. PK converts phosphoenolpyruvate to lactate, leading to the generation of ATP through the Embden-Meyerhof pathway.130 Thus, PK-deficiency causes decreased ATP levels.130 PK-deficiency is inherited as an autosomal recessive disease and common in specific areas.31 However, the exact prevalence of PK-deficiency in Korea is currently unknown.
There are some limitations to this study. 1) Not all administrative districts of Korea were covered because this study involved a questionnaire-based survey. Because there are no pediatric hematologists in Sejong City, this area was not included in the survey. The HHA patients in this city are diagnosed in other cities or provinces. 2) The recruitment hospitals were not completely identical between the 1997–2006 study and the 2007–2016 study due to changes in the faculty's overseas training, personnel transfers, or lack of response. 3) In the previous 1997–2006 study, data regarding genetic information of HHA or ethnicity of parents had not been collected. Therefore, we could not directly compare the genetic information or number of foreign parents between the 1997–2006 study and the 2007–2016 study.
In conclusion, Korean HHA patients could be successfully diagnosed according to Korean SOP established by RBC Disorder WP of KSH, and the proportion of newly diagnosed hemoglobinopathies and RBC enzymopathies have significantly increased in the recent Korean pediatric HHA cohort (2007–2016), when compared to the previous Korean pediatric HHA cohort (1997–2006). The increase in hemoglobinopathies is most probably due to the increase in interracial marriages between Korean male and South-East Asian female and increase in awareness among clinicians of the possibility of HHA in Korean pediatric anemia patients. The increase in enzymopathies, especially PK deficiency, is believed to be due to increased availability of genetic diagnostic techniques. Nonetheless, 6.5% of HHA patients still do not have a clear diagnosis. Therefore, for HHA patients whose diagnosis cannot be made by the usual methods, it is necessary to improve access to genetic tests such as next-generation sequencing (NGS) at reasonable costs. Genetic testing for HHA might help in determining novel mutations that have been introduced into the Korean population as a result of immigration. It is necessary to continue epidemiologic research of HHA every 10 years and to increase diagnostic value of HHA with the introduction of advanced diagnostic technology including NGS panel in the SOP of HHA in near future.
 XML Download
XML Download