

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
The deleterious effects of ozone (O3) exposure on human health, particularly the lungs, have been observed in humans and demonstrated in animal experiments. Exposure to high levels of O3 is associated with higher rates of emergency room visits, hospital admissions of patients with asthma and chronic obstructive pulmonary disease,123 asthma, and rhinitis.45 In addition, airway inflammation and epithelial damage are apparently induced in experimental animals exposed to O3.6789 One mechanism involved is the overproduction of reactive oxygen species (ROS), which can act as a signaling factor to induce inflammation.10 ROS also induce lipid peroxidation of cell membranes and the oxidation of amino acids to inactivate membrane-bound receptors.1112 This damage may modify the functions of signal transduction molecules as well as membrane molecules, such as cystic fibrosis transmembrane conductance regulator (CFTR); membrane-spanning 4-domains, subfamily A, member 4C (Ms4a4c); interferon-induced transmembrane protein 6 (Ifitm6); transmembrane protein 37 (Tmem37); and solute carrier family 26 member 4 (Slc26a4; pendrin).13 In fact, CFTR protein and mRNA expression and chloride currents in cultured human bronchial epithelial cells are decreased by O3 exposure.14 The transport of anions such as Cl− and HCO3− in the airway epithelium is important for controlling electrolytes and fluid secretion and pH regulation. Cl− transport in particular is recognized as one of the most important factors in the regulation of airway surface hydration, and therefore in mucociliary clearance.15 This transport system is defective in cystic fibrosis, in which the function of CFTR is lacking.16 By contrast, Slc26a4 is a Na+-independent antiporter exchanger for anions such as Cl− and thiocyanate (SCN−).17 Slc26a4 regulates the airway surface liquid volume via Cl− and subsequent water absorption and excretion of HCO3−, increasing the viscosity of airway-lining fluids.17 This protein is involved in the rhinoviral infection-induced exacerbation of asthma,1819 allergen-induced exacerbations,20 and bacterial infections,21 as well as allergic rhinitis and chronic rhinosinusitis.22 Forced expression of Slc26a4 in NCI-H292 cells or mouse airway epithelia induces goblet cell hyperplasia (GCH), airway inflammation, and hyperreactivity independent of T helper 2 (Th2) cell responses.23 In addition, SCN−, a metabolite regulated by Slc26a4, plays an antioxidant role in the pathogenesis of inflammation-related diseases.24 Thus, the regulation of Slc26a4 expression is an optimal strategy against airway inflammation following exposure to air pollutants such as O3.
Slc26a4 expression in renal tubules is strongly enhanced during metabolic alkalosis and markedly reduced by the intake of acids such as ammonium chloride (NH4Cl).2526272829 However, few studies have investigated the role of Slc26a4 in O3-induced airway injury and the potential therapeutic effects of treatment with acids such as NH4Cl. In the present study, we measured the expression levels of Slc26a4 protein and mRNA in the lungs of mice after O3 inhalation and evaluated the therapeutic effects of NH4Cl on O3-induced airway injury.
METHODS
Protocol of O3 exposure and NH4Cl treatment in mice
Female 6-week-old BALB/c mice (Orient Bio, Inc., Seongnam, Korea) were individually housed in rack-mounted stainless steel cages under pathogen-free conditions with free access to food and water. The mice were exposed to filtered air or 2 ppm O3 in whole-body exposure chambers 3 hours daily for 21 days as described previously.67 O3 was generated using a Sander Model 50 ozonizer (Erwin Sander Elektroapparatebau GmbH, Uetze-Eltze, Germany) and the levels within the chambers were monitored using an ambient air O3 analyzer (Model 49C; Thermo Environmental Instruments Inc., Franklin, MA, USA). Air-sampling probes were placed in the breathing zones of the mice. The O3 concentration (mean ± standard error of the mean [SEM]) in the chamber was 1.98 ± 0.06 ppm during the exposure period. Sham mice were challenged with filtered air with 0.2 ± 0.01 ppm O3 concentration. Mice were administered 50 μL NH4Cl (0.1, 1, 10 mM; Sigma-Aldrich, St. Louis, MO, USA) in phosphate-buffered saline intranasally 5 min before O3 exposure on Days 7, 9, 11, 13, 15, 17, 19, and 21 as described in Supplementary Fig. 1.
Measurement of lung resistance
Lung resistance was measured on Day 21. The mice were anesthetized with ketamine–xylazine. The trachea was excised, cannulated with a polyethylene tube, and mechanically ventilated using the flexiVent system (SCIREQ Inc., Montreal, Canada) as previously described.3031 Increasing concentrations of aerosolized methacholine (0, 5, 20, and 100 mg/mL, Sigma-Aldrich) were sequentially inhaled for 3 minutes, and lung resistance was measured.
Procedure of bronchoalveolar lavage (BAL) and sample preparation
BAL was performed through the tracheal tubes immediately after measuring lung resistance. The mice were sacrificed by exsanguination from the abdominal aorta. Lungs were lavaged four times with 1.0 mL physiologic saline (4.0 mL in total). The bronchoalveolar lavage fluid (BALF) was filtered through 4 × 4 gauze and centrifuged at 500 ×g for 5 minutes. The cell pellet was immediately suspended in 4 mL physiologic saline, and total cell numbers were counted in duplicate with a hemocytometer (Neubauer counting chamber) with Trypan blue dye exclusion for viability. A 200 µL aliquot was used to prepare slides for differential cell counts and immunocytostaining by cytocentrifugation (Model 2 Cytospin; Shandon Scientific Co., Pittsburgh, PA, USA). Differential cell counts were made on slides stained with Diff-quick, and 500 or more cells were counted per slide at 400× magnification.
Histological analyses and western blotting
The left lungs and trachea were filled intratracheally with fixative (0.8% formalin, 4% acetic acid). Then the left lungs were removed and fixed with 10% (vol/vol) neutral buffered formalin. The specimens were dehydrated and embedded in paraffin. The tissues were cut into slices 4 µm thick for hematoxylin & eosin (H&E), periodic acid-Schiff (PAS), and immunofluorescence (IF) staining for Slc26a4 and mucin 5AC (Muc5ac). The tissue sections were de-paraffinized, re-hydrolyzed, and blocked with normal serum for 1 hour at room temperature. Peribronchial inflammation was semi-quantitatively graded for cellular infiltration around airways as follows: normal (grade 0), few cells (grade 1), one-cell-deep ring of inflammatory cells (grade 2), or more than a four-cells-deep ring of inflammatory cells (grade 3) on H&E-stained lung slices under light microscopy.3233 Goblet cells were counted as the number of PAS-positive cells among the total number of bronchial epithelial cells.7 All measurements were performed using Image J software (National Institutes of Health, Bethesda, MD, USA) by two examiners in a blinded fashion and averaged. For IF staining, the slices were incubated at 4°C with rabbit anti-mouse Slc26a4 polyclonal antibody (1:200 dilution; Abcam, Cambridge, UK) and goat anti-mouse Muc5ac (K-20) polyclonal antibody (1:200 dilution; Abcam). After washing three times in Tris-buffered saline (TBS), the slides were incubated for 1 h at room temperature with fluorescein isothiocyanate (FITC)-conjugated anti-rabbit immunoglobulin G (IgG) and phycoerythrin (PE)-conjugated anti-goat IgG (1:1,000 dilution; Abcam) diluted in normal serum. After washing in TBS, the slides were incubated with 4′,6-diamidino-2-phenylindole (DAPI) (1:1,000 dilution; Sigma-Aldrich). The slides were washed in TBS, mounted, and observed under a confocal laser-scanning microscope (CLSM) (LSM 510 META; Zeiss, Jena, Germany).
A portion of right lung was treated with radioimmunoprecipitation assay (RIPA) buffer (Scientific Products, Gibbstown, NJ, USA) containing 0.5 mM EDTA and 100 mM phenylmethylsulfonyl fluoride (PMSF), and stored at −80°C for western blot analyses. The extracted lung tissues were homogenized in a protein lysis solution containing 50 mM Tris-HCl (pH 7.4), 50 mM NaCl, 0.1% sodium dodecyl sulfate (SDS), and 1% Triton X-100 in distilled water, then centrifuged at 14,000 rpm for 30 minutes at 4°C. Then 30 µg of the soluble portion was loaded and electrophoresed with a discontinuous system consisting of a 7.5% and 15% polyacrylamide gel. The proteins were transferred to a nitrocellulose membrane at 60 V for 2 hours. The membrane was blocked using 5% skim milk containing 0.1% Tween-20 made in TBS-T for 1 hour at room temperature and incubated overnight with rabbit polyclonal anti-Slc26a4 (1:300; Abcam), mouse monoclonal anti-caspase-1 (1:1,000; Adipogen, San Diego, CA, USA), mouse monoclonal anti-interleukin (IL)-1β (1:1,000; Cell Signaling Technology, Beverly, MA, USA), rat monoclonal anti-interferon gamma (IFN-γ) (1:1,000; BD Biosciences, San Jose, CA, USA), mouse monoclonal anti-tumor necrosis factor alpha (TNF-α) (1:1,000; Abcam), rabbit polyclonal anti-phospho-nuclear factor kappa B (NF-κB) (1:1,000; Cell Signaling Technology), rat monoclonal anti-IL-17 (1:1,000; R&D Systems, Inc., Minneapolis, MN, USA), or mouse monoclonal anti-β-actin (1:5,000; Sigma-Aldrich) overnight at 4°C. Then they were incubated for 1 hour at room temperature with a horseradish peroxidase-conjugated secondary antibody (1:5,000; Santa Cruz Biotechnology, Santa Cruz, CA, USA). The target protein was detected using an enhanced chemiluminescence solution (Amersham Pharmacia Biotech, Little Chalfont, UK) on X-ray film.
Real-time polymerase chain reaction (PCR) with Slc26a4 mRNA
Total RNA was extracted from the lungs using TRIzol reagent (Ambion, Carlsbad, CA, USA) according to the manufacturer's recommendations. The cDNA was synthesized from 3 µg total RNA using a Superscript II kit (Invitrogen, Carlsbad, CA, USA) and amplified by PCR. Real-time PCR was performed using the StepOne Real-Time PCR System (Applied Biosystems, Foster City, CA, USA). The PCR mixture (20 μL) contained 1 μg cDNA, 1 μL 10 pmol forward (AAGAGAGCCTTTGGTGTGGTA) and reverse (CAGGGCATAAGCCATCCCTTG) primers, and 10 μL 2× Power SYBR Green PCR Master Mix (Applied Biosystems). The reaction was carried out in a two-step procedure: denaturation at 95°C for 15 seconds and 60°C for 1 minute, then cycles of melting at 95°C for 15 seconds, 60°C for 1 minute, and 95°C for 15 second. Data were analyzed using the 2-ΔΔCT method and are presented as the relative fold-change after normalization to peptidylprolyl isomerase A (PPIA).34
Measurement of SCN−
SCN− was measured spectrophotometrically in BALF and lung lysates as described previously, with slight modifications.35 Trichloroacetic acid (TCA) was added to standards (Sigma-Aldrich) or 100 µL samples in microcentrifuge tubes to a final concentration of 5% TCA. Then the standards or samples were transferred to a 96-well plate in duplicate with 50 μL per well, then 50 μL chlorinating reagent (197 mM Na2HPO4 and 0.6% [v/v] NaOCl) was added. Immediately thereafter, 30 µL colorimetric reagent (463 mM NaOH, 224 mM 1,3-dimethylbarbituric acid and 232 mM isonicotinic acid) was added. The plate was incubated in the dark for 5 minutes, then the absorbance was measured at 607 nm using a SpectraMax 340PC plate reader (Molecular Devices, San Jose, CA, USA).
Statistical analyses
The data were analyzed using SPSS ver. 20.0 software (IBM Corp., Armonk, NY, USA). Comparison of variables was performed using analysis of variance, and the Tukey's honestly significant difference test was applied as a post hoc analyses. The data are presented as the mean ± SEM. P < 0.05 was considered statistically significant.
RESULTS
Temporal changes in Slc26a4 gene and protein expression in mice following O3 exposure
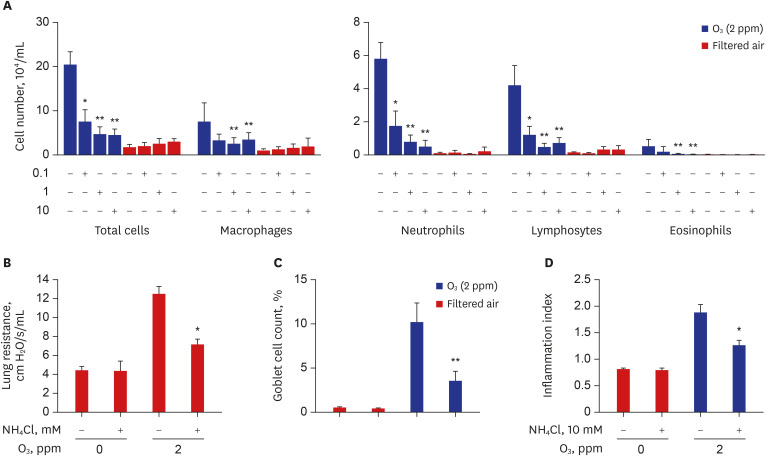
During exposure to 2 ppm O3, the numbers of total cells, neutrophils, macrophages, and lymphocytes in BALF were significantly increased from Days 3 to 7 compared to those of Day 0 (P < 0.05), and the levels peaked at Day 21 (Fig. 1A). The peribronchial inflammation index showed similar changes. Compared to Day 0, O3 exposure increased cellular infiltration around the bronchi, bronchioles, and alveoli at Day 21 (Fig. 1B and C). Concomitantly, goblet cell percentages were significantly increased at Day 7 and maximal at Day 21 (P < 0.05 and P < 0.01, respectively) (Fig. 1B). Slc26a4 mRNA (Fig. 2A) and protein levels (Fig. 2B) were elevated at Day 1 and progressively increased through Day 21 in a time-dependent manner. SCN− levels also increased in BALF and lung tissues at Day 1 and were persistently elevated through Day 21 compared to the levels at Day 0 (P < 0.05) (Fig. 2A).
Fig. 1
Temporal changes in inflammation profiles and goblet cells in mice exposed to O3 (2 ppm for 3 hr/day). (A) Inflammatory cell profiles in bronchoalveolar lavage fluid (BALF). (B) Number of goblet cells, and score of the inflammation index. Data represent the mean ± SEM of six experiments. P values were calculated by ANOVA and t-tests. (C) Representative images of H&E-stained lung tissues in O3-exposed mice.
O3 = ozone, SEM = standard error of the mean, ANOVA = analysis of variance, H&E = hematoxylin & eosin.
*P < 0.05 compared to 0 hours; **P < 0.01 compared to 0 hours.

Fig. 2
Temporal changes in Slc26a4 (pendrin) and SCN− levels in mice exposed to O3 (2 ppm for 3 hr/day). (A) Levels of SCN− in BALF and lung lysates and Slc26a4 mRNA levels normalized to PPIA in lung lysates after exposure to 2 ppm O3. Data represent the mean ± SEM of six experiments and P values were determined by t-test. (B) Western blot of Slc26a4 levels in lung lysates pooled from 3 mice after exposure to 2 ppm O3.
PPIA = peptidylprolyl isomerase A, Slc26a4 = solute carrier family 26 member 4, SCN− = thiocyanate, BALF = bronchoalveolar lavage fluid, SEM = standard error of the mean.
*P < 0.05 compared to 0 hours.

Effects of NH4Cl on O3-induced inflammation, airway resistance, and goblet cells in mice
During exposure to 2 ppm O3 through Day 21, NH4Cl was administered every other day from Day 7 to Day 21. BALF and lung tissues were obtained on Day 21 (Supplementary Fig. 1). NH4Cl attenuated the increased numbers of total cells, macrophages, neutrophils, and lymphocytes in O3-exposed mice in a dose-dependent manner with 0.1, 1, and 10 mM NH4Cl (Fig. 3A). Treatment with 10 mM NH4Cl significantly decreased the enhanced lung resistance of the O3-exposed mice (Fig. 3B) with concomitant attenuation of the increased goblet cell number and the increased peribronchial inflammation index (Fig. 3C, D and Supplementary Fig. 2).
Fig. 3
Effects of NH4Cl on inflammation and resistance in the lungs of mice exposed to O3 (2 ppm for 3 hr/day). (A) Effects of NH4Cl on the numbers of total cells, macrophages, neutrophils, eosinophils, and lymphocytes in BALF of mice exposed to O3 (2 ppm for 3 hr/day) for 21 days. (B) RL (cm H2O/s/mL) measured using the flexiVent system with increasing concentrations of methacholine and represented as the area under the curve of RL. (C) Effects of NH4Cl on the number of goblet cells and (D) inflammation index score in O3-exposed mice. Data represent the mean ± SEM of six experiments. P values were calculated using ANOVA and t-tests.
NH4Cl = ammonium chloride, O3 = ozone, BALF = bronchoalveolar lavage fluid, RL = lung resistance, SEM = standard error of the mean, ANOVA = analysis of variance.
*P < 0.05 compared to untreated mice (0 mM); **P < 0.001 compared to untreated mice (0 mM).
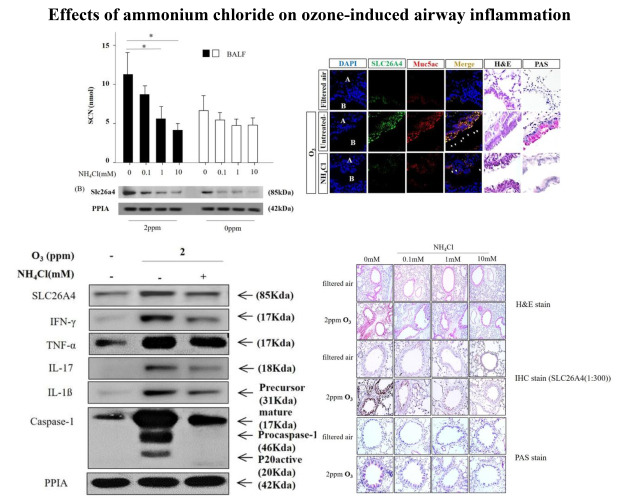
Effects of NH4Cl on the O3-induced Slc26a4 expression and SCN− levels
Western blot analyses demonstrated that treatment with NH4Cl decreased Slc26a4 protein levels in lung lysates in a dose-dependent manner, with a concomitant decrease in SCN− levels in BALF (Fig. 4A and B). In IF staining, Slc26a4 and Muc5ac double-positive cells were increased in the bronchial epithelium of O3-exposed mice at Day 21, and treatment with 10 mM NH4Cl attenuated the increased expression of Slc26a4 and Muc5ac toward the levels observed in filtered air-treated mice (Fig. 4C).
Fig. 4
Localization of Slc26a4 and temporal changes in Slc26a4 and SCN− levels in O3-exposed mice, and the inhibitory effects of NH4Cl. (A) Representative SCN− in mice BALF of sham, O3-exposed, and NH4Cl-treated O3-exposed mice on Day 21. (B) Representative western blot of Slc26a4 in lung lysates pooled from sham (n = 6), O3-exposed (n = 6), and NH4Cl-treated O3-exposed (n = 6) mice on Day 21. Data represent the mean ± SEM of six experiments and P values were determined using t-tests. (C) Representative image of H&E and immunofluorescence staining for Slc26a4 (green, FITC) and Muc5ac (red, PE) in lung tissues of sham, O3-exposed, and NH4Cl-treated O3-exposed mice on Day 21. Nuclei were stained with DAPI (blue) (1,000×).
Slc26a4 = solute carrier family 26 member 4, SCN− = thiocyanate, O3 = ozone, NH4Cl = ammonium chloride, SEM = standard error of the mean, H&E = hematoxylin & eosin, Muc5ac = mucin 5AC, DAPI = 4′,6-diamidino-2-phenylindole, A = alveoli, B = bronchiole.
*P < 0.05 compared to controls.

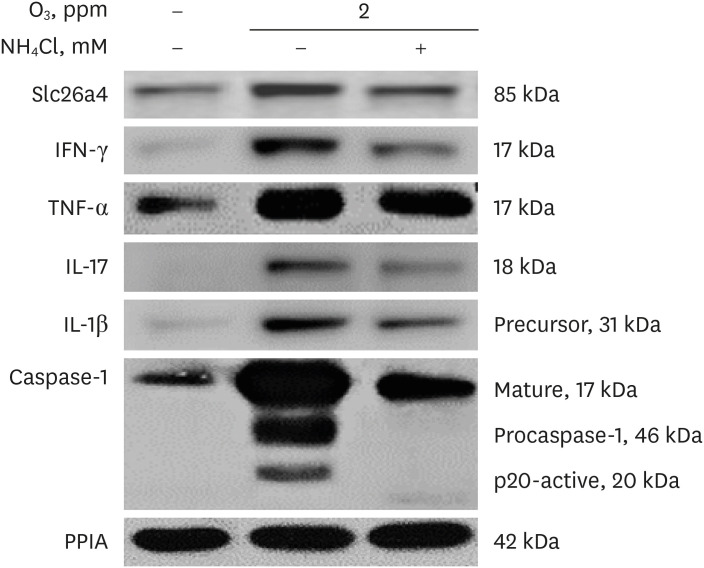
Western blot of Th1 and Th17 cytokines and inflammatory mediators
In western blots, Slc26a4 protein levels peaked on Day 21 in pooled lung lysates of the 2 ppm O3-exposed mice. Concomitantly, levels of IFN-γ, TNF-α, IL-17, mature forms of IL-1β, and p20-activated caspase-1 were increased in the O3-exposed mice. The increased levels of these cytokines were markedly downregulated by treatment with 10 mM NH4Cl (Fig. 5). Ozone-induced Slc26a4 upregulation may induce intracellular transport of Cl− and extracellular transport of HCO3−, leading to a reduction in airway surface liquid, mucus thickening, and impaired mucociliary clearance.
Fig. 5
Effects of Slc26a4 on cytokine levels. Representative western blot of Slc26a4 and neutrophil-related cytokine levels in lung lysates pooled from O3-exposed mice (n = 6, O3 dose 2 ppm) and 10 mM NH4Cl-treated mice on Day 21 (n = 6, O3 dose 0.2, 2 ppm). Temporal changes in Slc26a4, IFN-γ, TNF-α, IL-1β, P20-activated caspase-1, and IL-17 levels in O3-exposed mice, and the inhibitory effects of NH4Cl.
Slc26a4 = solute carrier family 26 member 4, O3 = ozone, NH4Cl = ammonium chloride, IFN-γ = interferon gamma, TNF-α = tumor necrosis factor alpha, IL = interleukin, PPIA = peptidylprolyl isomerase A.
DISCUSSION
We demonstrated that ozone exposure significantly increased Slc26a4 protein and mRNA levels, with concomitant increases in epithelial Muc5Ac protein expression and SCN− levels in BALF. These results indicate that O3-induced GCH and airway inflammation are accompanied by upregulation of Slc26a4 with functional activation to elevate SCN− levels inside airways. We did not measure changes in CFTR in the present study; however, ozone stress downregulates CFTR protein and mRNA expression and chloride current.15 Thus, the increase in Slc26a4 and the loss of CFTR function exacerbated excessive fluid absorption, with a lack of fluid secretion leading to further reduction of watery components in the airway surface liquid in ozone-exposed conditions.
Slc26a4 participates in the development of several respiratory and allergic diseases, including rhinoviral infection-induced exacerbation of asthma,1819 allergen-induced exacerbations,2021 and allergic rhinitis and chronic rhinosinusitis,22 which are accompanied by excessive mucus production. In this process, Slc26a4 may be a common mediator for mucus production. Mouse models of asthma and chronic obstructive lung disease exhibit upregulation of Slc26a4 and consequently increased levels of Muc5ac, a major component of airway mucus.2123 In the present study, Muc5ac and Slc26a4 were co-expressed in bronchial epithelium, mainly at apical sites, which indicates that Slc26a4 is mainly expressed in goblet cells.
GCH, a feature of asthma and other respiratory diseases, is driven by the Th2 cytokines IL-4 and IL-13. IL-4 also induces the expression of many other genes encoding ion channels and transporters, including TMEM16A, Slc26a4, SLC12A2, and ATP12A. IL-4 enhances calcium- and cAMP-activated chloride/bicarbonate secretion, resulting in high bicarbonate concentrations and alkaline pH in the fluid covering the apical surface of epithelia.36 In addition, Slc26a4 is upregulated by several cytokines, including IL-13,203738 IFN-γ, and IL-17A.18 In the present study, IL-17 and IFN-γ levels significantly increased in O3-exposed mice lung lysates. Thus, IL-17 and IFN-γ may be the primary cytokines inducing Slc26a4 expression in O3-exposed mice. Furthermore, IFN-γ and IL-17A released from the activated Th1 and Th17 lymphocytes were correlated with O3-induced airway inflammation and GCH.394041 Neutrophils were present in large numbers in the airways of the O3-exposed mice in the present study. In the western blot analyses, p20-activated caspase-1, IL-1β, IFN-γ, and IL-17 levels were elevated on Day 21 after O3 exposure. Our data suggest that Slc26a4 may be related to the activation of the inflammasome and the production of IFN-γ and IL-17A. Because we did not use transgenic or knockout mice, there are limitations in interpreting our results, and we cannot ascertain whether the elevation of Slc26a4 protein is the cause, result, or a parallel phenomenon with the elevation of the neutrophil-activated cytokines.
In our in vivo animal studies, blocking Slc26a4 with NH4Cl attenuated neutrophilic inflammation, enhanced airway reactivity, and GCH. To the best of our knowledge, this is the first study demonstrating inhibitory effects of NH4Cl on O3-induced airway injury in mice. Regulation of Scl26a4 by NH4Cl loading is well known in the kidney. NH4Cl loading (0.033 mmol NH4Cl per g body weight for 7 days) dramatically reduces Scl26a4 abundance in intercalated cells of the cortical collecting duct and connecting tubule.2526272829 NH4Cl was administered from Days 7 to 21 because Slc26a4 began to appear at Day 7 in the western blot analyses, and increasing the concentration of NH4Cl substantially restored the increases in neutrophil numbers and Slc26a4 levels to those of the sham-treated mice in a dose-dependent manner, with a concomitant decrease in airway hyperreactivity. The treatment concomitantly attenuated the elevated IL-1β, IFN-γ, and IL-17 levels. NH4Cl treatment also suppressed the number of lymphocytes in the O3-exposed mice. Although we did not assess Th1 and Th17 subsets using these lymphocytes in BALF, they may be the sources, which will be resolved in a future study.
Glutathione and SCN− are two thiols that protect against oxidative injury.35 In the present study, SCN− was measured in BALF and lung lysates. SCN− transport is carried out in bronchial epithelial cells through the mechanisms of CFTR, upregulation of calcium-dependent Cl− channels, and Slc26a4.1718 Interestingly, SCN− levels were elevated in BALF but not significantly higher in lung tissues. In the IF-stained lung tissues, Slc26a4 was dominantly expressed in bronchial epithelium. Thus, O3 may induce Slc26a4 protein expression in bronchial epithelial cells, and the enhanced Slc26a4 function may lead to increased SCN− levels inside airways. The main limitation of the present study was the absence of experiments using Slc26a4 transgenic or knockout mice for clear determination of the contribution of activated Slc26a4 gene and protein in O3-exposed mice. Another limitation was the use of NH4Cl as a Slc26a4 inhibitor, rather than using specific inhibitors for Slc26a4 such as anti-sense microRNAs. Additionally, NH4Cl may be reacting with O3 and produced the nitric oxide (NO), 2H2O and Cl− (BALANCING REDOX REACTIONS; https://www.periodni.com/half-reaction_method.php). NO is a signaling molecule that plays a key role in the pathogenesis of inflammation. NO is considered as a pro-inflammatory mediator that induces inflammation due to over production in abnormal situations.
In conclusion, Slc26a4 protein levels were markedly increased in the bronchial epithelium of O3-exposed mice and were significantly correlated with airway neutrophils and GCH, with concomitant elevation of IL-1β, IL-17, and IFN-γ levels. Blocking Slc26a4 with NH4Cl restored neutrophilic inflammation and increased airway resistance to the levels of sham-treated mice. These data indicate that Slc26a4 may initiate and amplify the neutrophilic inflammation of asthma directly or in collaboration with elevated IL-1β, IL-17, and IFN-γ. The modulation of Slc26a4 protein levels in the airway may provide a therapeutic strategy to control neutrophilic inflammation in lung diseases characterized by Slc26a4 overexpression.
 XML Download
XML Download