

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Haploinsufficiency of A20 (HA20) is a rare hereditary autoinflammatory syndrome caused by mutations in the TNFAIP3 gene, which encodes the protein A20 (or TNAP 3).12 This condition is a newly recognized nuclear factor (NF)-κB-mediated autoinflammatory disease; it is also called autosomal dominant familial Behçet's disease. The protein A20 is a potent inhibitor of the NF-κB signaling pathway that restricts NF-κB signals via deubiquitinating enzyme (DUB) activity. Cells expressing the mutant A20 protein exhibit an impaired mechanism for the removal of K63-linked ubiquitin from TRAF6, NEMO, and RIP1 proteins after tumor necrosis factor (TNF) stimulation. HA20 causes activation of the NF-κB pathway, leading to an increased proinflammatory cytokine expression and systemic inflammation.23
The first description of mutations in the TNFAIP3 gene involved in NF-κB pathway regulation was reported by Zhou et al. in 2016.1 Here, we report the first case of HA20 in Korea, which was initially suspected as neonatal lupus erythematosus (NLE).
Go to : 
CASE DESCRIPTION
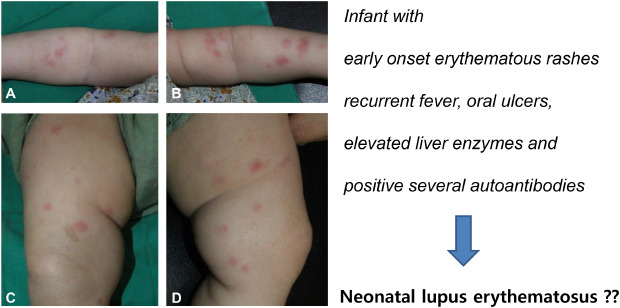
A 2-month-old infant suffering from recurrent fever and rash on both cheeks was admitted to another hospital. She showed elevated inflammatory markers and liver enzymes and was treated with antibiotics, but the fever did not subside. She was referred to our hospital after a month (at 3 months of age). On admission, a physical examination revealed persistent erythematous wheal-like patches and café au lait spots on her entire body (Fig. 1). The rash initially appeared on her face and then spread to her trunk and limbs. Small ulcers were observed on her palate and buccal mucosa. Initial laboratory test findings and follow-up data are summarized in Tables 1 and 2.
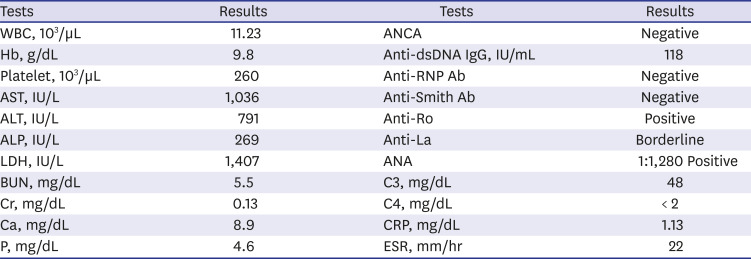
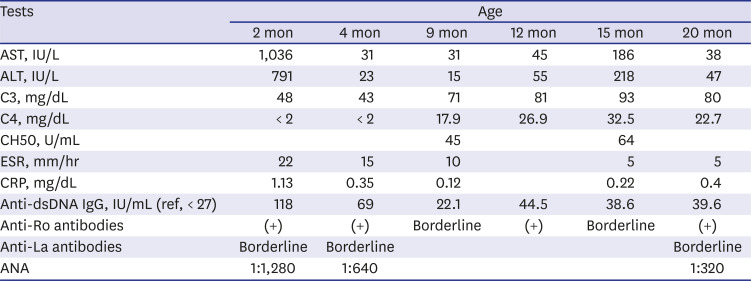
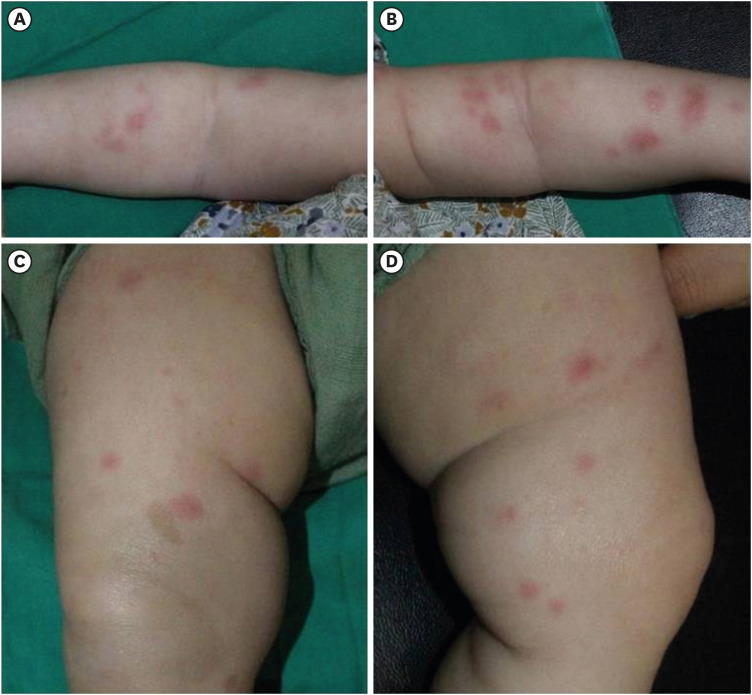 | Fig. 1Figures show erythematous wheal-like patches on the whole body and 4 extremities (A-D). You can also see the café au lait spot on (C). Figures are published under Informed consent.
|
Table 1
Major results of initial laboratory tests
This table shows the major results of initial laboratory tests.
WBC = white blood cells, Hb = hemoglobin, AST = aspartate transaminase, ALT = alanine transaminase, ALP = alkaline phosphatase, LDH = lactate dehydrogenase, BUN = blood urea nitrogen, Cr = creatinine, Ca = calcium, P = phosphorus, ANCA = anti-neutrophil cytoplasmic antibody, dsDNA = double strand DNA, IgG = immunoglobulin G, RNP = ribonucleoprotein, Ab = antibody, ANA = antinuclear antibody, CRP = C-reactive protein, ESR = erythrocyte sedimentation rate.
![]()
Table 2
Follow-up results
AST = aspartate transaminase, ALT = alanine transaminase, ESR = erythrocyte sedimentation rate, CRP = C-reactive protein, dsDNA = double strand DNA, IgG = immunoglobulin G, ANA = antinuclear antibody.
![]()
At first, we suspected NLE because of the typical rashes observed and positive autoantibodies, including antinuclear antibodies, anti-double stranded DNA antibodies, anti-Ro antibodies, and anti-La antibodies. However, her mother had no history of systemic lupus erythematosus (SLE) or Sjogren's syndrome. Several laboratory tests were performed to identify autoantibodies in the mother; these were all negative. The infant's fever and rash abated a little without specific treatment; regular follow-up was maintained on an outpatient basis. The infant's development was normal, but her height was approximately three percentiles, and weight was around 25 percentiles. At 7 months of age, her skin rashes became distinct and began to spread again. A skin biopsy was performed, which demonstrated multifocal histiocytic infiltration, mainly in the perivascular and periappendiceal areas in the dermis and subcutis suggesting panniculitis. Levels of C3 and C4 improved, but tests for autoantibodies were still positive. Then, we considered the possibility of other diseases that could cause early onset rashes and abnormal autoantibodies, including autoinflammatory syndrome, monogenic SLE, or complement deficiency, all of which are rare. Reexamination of detailed family history revealed that her father had recurrent symptoms, including oral and genital ulcers, knee arthralgia, abdominal pain, and diarrhea. These Behçet-like symptoms have lasted for many years, since he was a teenager, and he takes medications irregularly when they are severe but does not want the full-scale treatment. Her grandmother also had suffered from long-standing ankylosing spondylitis-like disease, although we could not check the details of the disease.
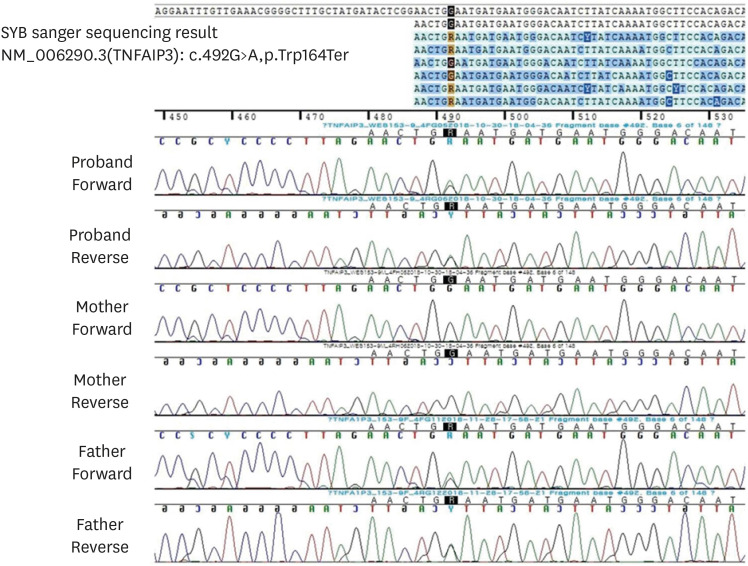
Whole-exome sequencing (WES) was conducted to identify a possible genetic disorder, which revealed a nonsense mutation in the TNFAIP3 gene, leading to HA20. The variant was confirmed by Sanger sequencing in the infant and her father. Although it is a novel mutation, considering that it is a nonsense mutation leading to early termination of the protein A20 and a truncated protein during translation, it is a definite pathogenic mutation (pathogenic predictions by MutationTaster revealed a pathogenic effect). Moreover, it has already been proven that this gene acts as a haploinsufficiency, so we believe that no additional functional study is necessary to confirm its pathogenicity.12 Also, the infant's and her father's symptoms point towards an autoinflammatory syndrome called autosomal dominant Behçet's disease, caused by mutations in the TNFAIP3 gene. Additionally, WES revealed another known pathogenic mutation in the NF1 gene (c.4103T>G, p.Leu1368*, heterozygote), causing neurofibromatosis, which was evident due to the presence of multiple café au lait spots. Subsequently, the fever and rashes gradually diminished, and she is currently followed up without routine medications. Her latest laboratory tests (Table 1) still showed positive results for some autoantibodies. During her follow-up, she was sometimes admitted at local hospitals because of intermittent severe oral ulcers and poor feeding. Rashes occasionally occurred but disappeared mostly within one week. A TNF blocker would be administered in case her symptoms worsened.
Methods and results of WES
The SureSelect Human All Exon V5 kit (Agilent Technologies, Santa Clara, CA, USA) was used for library preparation, and sequencing was performed on the Illumina NextSeq500 platform (Illumina Inc., San Diego, CA, USA), generating 2 × 150 bp paired-end reads. Alignment of sequence reads, indexing of the reference genome (hg19), and variant calling with a pipeline based on GATK Best Practice was performed. Alignment was carried out using BWA-mem software (version 0.7.12),4 and duplicated reads were marked using Picard software (version 1.96, http://picard.sourceforge.net). Local alignment, base quality recalibration, and variant calling was performed using the Genome Analysis Tool kit (GATK, version 3.5), and the annotation was done using VEP88 (Variant Effect Predictor) and dbNSFP v3.3 software.567 The mean depth of coverage was 103× and approximately 98% of the targeted bases were read more than 10×. Using population databases—Exome Variant Server (http://evs.gs.washington.edu/EVS/), Exome Aggregation Consortium (http://exac.broadinstitute.org/), gnomAD (http://gnomad.broadinstitute.org/)—and a population specific database—Korean Reference Genome Database (http://152.99.75.168/KRGDB/menuPages/rstInfo.jsp), among the 480 variants with less than 1% of minor allele frequency, 130 variants were identified on genes associated with Mendelian diseases. After reviewing the variants with the phenotype of the patient, heterozygous nonsense variation of the TNFAIP3 gene was considered as the prime candidate; NM_006290.3:c.492G>A (p.Trp164Ter). This variation was neither found in population databases nor reported as a pathogenic variant in the literature or databases.
The DNA position appeared to be conserved across species (GERP++gt2 6.08; phyloP20way_mammalian 1.048; phastCons20way_mammalian 0.999). The variant was confirmed by Sanger sequencing in the infant and her father (Fig. 2).
Ethics statement
This study was approved by the Institutional Review Board in Pusan National University Yangsan Hospital (No. 05-2019-138) and we obtained informed consent from the parents for publication, including all figures.
Go to :
DISCUSSION
Autoinflammatory syndromes are generally characterized by recurrent attacks of unprovoked inflammation, without significant autoantibodies that represent innate immune system disorders. The term ‘systemic autoinflammatory disease’ was first proposed in 1999, followed by the identification of NLR inflammasomopathy, which is a well-known subtype.8910 Recently, several monogenic autoinflammatory syndromes related to interferon (interferonopathy) and the NF-κB pathway have been discovered along with inflammasomopathies. Autoinflammatory interferonopathies include systemic inflammation, panniculitis, and myositis due to proteasome defects (PRAAS/CANDLE) and vasculopathy, vasculitis, and interstitial lung disease with STING hyperactivity (SAVI).89
Protein A20 (or TNAP3), encoded by TNFAIP3, plays an important role in the negative regulation of inflammation and immune responses. HA20 is a newly discovered autoinflammatory disease, caused by loss-of-function mutations in TNFAIP3, which result in insufficient ubiquitin editing (DUB) activity of A20 and lead to increased NF-κB signaling and increased proinflammatory cytokine expression and systemic inflammation.211 The first series of familial early Behçet's disease cases associated with TNFAIP3 mutations were described in 2016.2
HA20 might manifest in patients as an early-onset, dominantly inherited disease, with a broad spectrum of autoinflammatory and autoimmune features. The clinical presentation may resemble Behçet's disease, juvenile idiopathic arthritis, inflammatory bowel disease, or periodic fever syndrome.3 The initial largest cohort of HA20 stated that the clinical appearance was heterogeneous, and the severity of the disease varied even within families, with some patients presenting with early-onset of severe symptoms whereas others exhibited late-onset of mild symptoms. All patients had recurrent painful ulcers in at least two sites, including oral (100%) and genital (94%). Other symptoms were arthritis (44%), bloody diarrhea (31%), recurrent fevers (50%), severe ocular inflammation (19%), and cutaneous manifestations (50%), ranging from pustular folliculitis-like rashes and acne to dermal abscesses.11 Our patient exhibited recurrent fever and rash, oral ulcers, and elevated liver enzymes but did not have arthritis, bloody diarrhea, or ocular inflammation yet. So far, her symptoms are like early-onset periodic fever syndrome than typical Behçet syndrome. However, as she grows up, other typical Behçet symptoms may become apparent.
A peculiar observation about HA20 is that, unlike other autoinflammatory syndromes, there may be some positive autoantibodies. Therefore, it may be difficult to distinguish this disease from other autoimmune diseases—SLE, Sjogren's syndrome, or, in the case of infants, neonatal LE. Aeschlimann and Laxer3 reported that half the patients had fluctuating amounts of autoantibodies, and two patients were initially diagnosed with SLE.11 Our patient also had fluctuating low autoantibodies from early infancy. The reason and mechanism for the development of these autoantibodies is not exactly known and needs further research. According to a few experimental studies, B cells or dendritic cells with tissue-specific deletion of A20 could lead to the production of autoantibodies.1213
NLE is an uncommon syndrome, which occurs due to transplacental passage of maternal autoantibodies (anti-Ro or anti-La). Its clinical presentation includes distinctive cutaneous lesions resembling those seen in SLE, liver disease, and cytopenia. However, most of these symptoms are reversed when the maternal autoantibodies are removed from the infant's blood, except a complete atrioventricular block, which is the most severe presentation and ultimately needs permanent pacemaker.14 Initially, we suspected NLE in our patient because of positive anti-Ro and anti-La antibodies and other clinical manifestations of NLE. However, she tested negative for maternal autoantibodies, which are critical for NLE diagnosis. Even six months after birth, the symptoms and autoantibodies persisted, leading us to an HA20 diagnosis after a detailed literature review. We performed WES because we could not rule out other autoinflammatory diseases and complement deficiencies. WES revealed likely pathogenic variant nonsense mutations in the TNFAIP3 gene leading to HA20 and a known pathogenic variant in the NF1 gene (c.4103T>G, p.Leu1368*, heterozygote). Two different genetic disorders can occur in the same patient and approximately 6% of the cases diagnosed using WES have two unrelated genetic diseases.15
Various drugs—colchicine, steroids, methotrexate, and thalidomide—are administrated to control this disease. Recently developed therapeutic approaches based on functional cytokine studies have shown that cytokine inhibitors—infliximab and anakinra—proved effective in suppressing systemic inflammation.1116
In conclusion, HA20 should be considered in the differential diagnosis of an infant with an early-onset dominantly inherited inflammatory disease that presents with recurrent oral and genital ulcerations and fluctuating autoantibodies. Additionally, it also should be considered in an infant with suspected NLE, whose symptoms and abnormal autoantibodies persist.
Go to :
 XML Download
XML Download