

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Heart failure has been a major cause of death worldwide for the past few decades. Arrhythmia, one of the causes of heart failure, is characterized by an abnormal rhythm of the heartbeat and is caused by dyssynchronous ion exchange or current in myocardial cells resulting from genetic mutations in protein channels.12 Prolonged persistence of arrhythmia can alter the excitation and contraction sequences to cause fibrillation3 and even death because blood cannot be ejected effectively.45 Short QT syndrome is one cause subject to arrhythmia due to genetic mutation in the ionic channel.
Short QT syndrome is described by the time interval of Q to T wave of the electrocardiogram (ECG) is less than 300 msec.6 The first identification of short QT syndrome was described by Gussak et al.7 The short QT syndrome increased arrhythmia generation because it shortened the refractory period, which prone to reentry resulting in chaotic electrical activity in the heart. Giustetto et al.8 stated that short QT syndrome is a rare condition because of a limited number of patients reported in published data, however, may play a role in sudden infant death. N588K KCNH2 is the first protein with a genetic mutation to be discovered in patients with short QT syndrome.69 In 2011, Zhang et al.10 performed an experimental study of L532P KCNH2 mutation which associates with short QT as the comparative subject with N588K mutation.
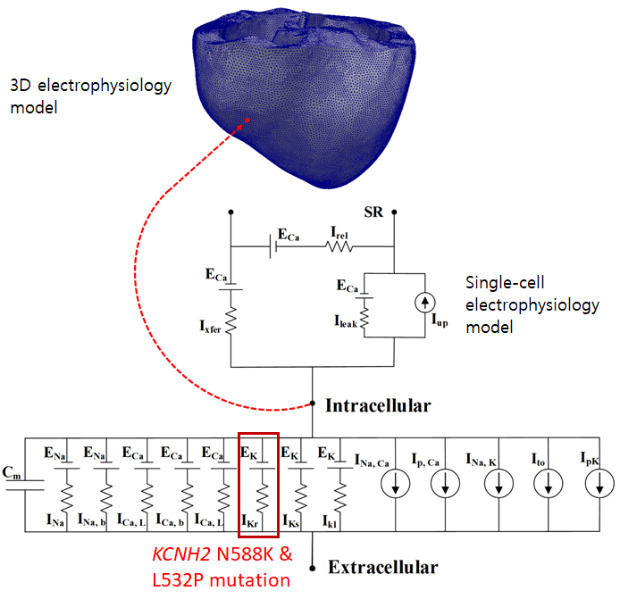
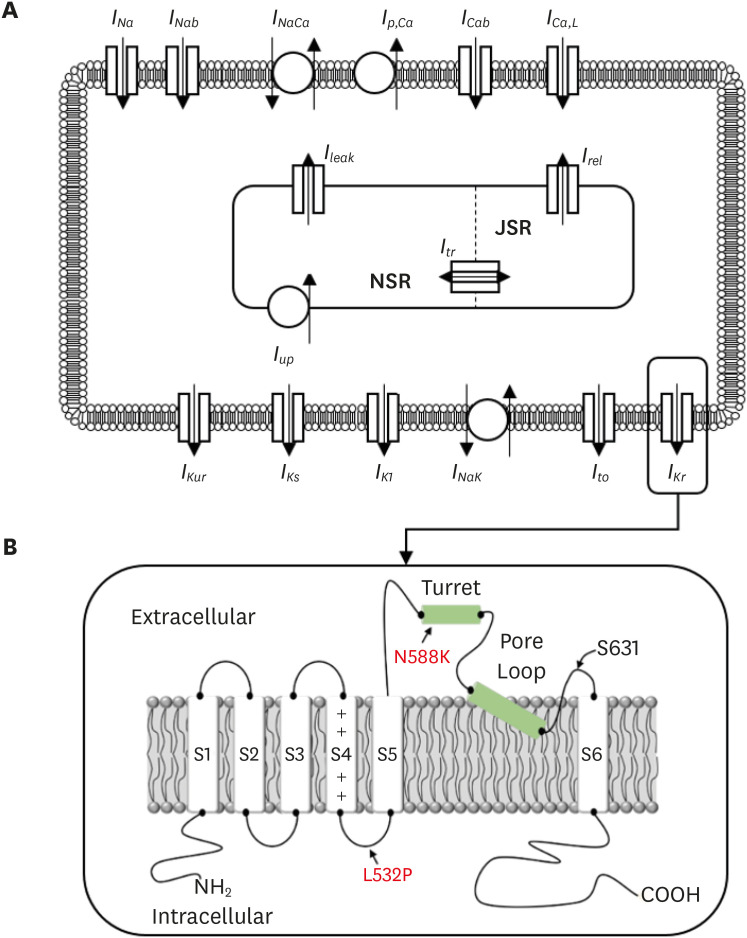
The KCNH2 mutation affects the alpha-subunit of the rapid delayed rectifier potassium channel (IKr) that plays a role in repolarization (Fig. 1A).61112 This gene can regulate changes in voltage activation in the heart and nervous tissue and is known to fasten cardiac repolarization by a gain of function and associated with short QT syndrome. N588K and L532P—the genetic mutations of KCNH2—are known to induce atrial arrhythmia. N588K is a fibril-related gain-of-function mutation in which the negatively charged asparagine (N) of residue N588 at the outer entrance of the KCNH2 channel subunit (S5) is replaced with the positively charged lysine (K) (Fig. 1B).9 In the L532P mutation, the leucine (L) of residue L532 located in the voltage sensor (S4) of the KCNH2 channel is replaced with proline (P) (Fig. 1B).13 L532P mutation is located in a different segment from the N588K in the KCNH2 and exhibited Kinect difference due to complex temperature dependency. Changes in the KCNH2 ion channel due to these mutations cause sudden cardiac death, resulting in abnormal QT intervals on the ECG.14 Therefore, it is essential to understand their association with cardiac arrhythmia through experiments related to KCNH2 ion channels.
The experimental measurement of KCNH2 ion channels is time-intensive and expensive. Hence, researchers have observed the effects of a few mutations on the action potential duration (APD) and the propagation signal in a simulation study using advanced cardiac electrophysiology in three-dimensional (3D) heart models. In 2012, Adeniran et al.15 performed a simulation to study the effect of the D172N mutation on the APD, ECG, and the electrical propagation signal in 3D ventricles. In 2014 and 2016, our group studied the relation of two mutations (V241F and G229D) with atrial arrhythmia using methods similar to those used by Adeniran et al.1617 In previous studies, simulations of the KCNH2 mutations—N588K and L532P—have been limited to a single cell model and a two-dimensional (2D) tissue model. Thus, quantification of the electric conduction patterns in the 3D heart geometry is limited. Therefore, in this study, we enhanced the observation of atrial conduction patterns due to KCNH2 ion channel mutation by applying the L532P and N588K mutations to the 3D atrium model as well. We hypothesize that L532P mutation plays a role in atrial fibrillation (AF) generation due to the significant shortening of APD by the mutation.
To simulate the AF conditions, we used Courtemanche-Ramirez-Nattel (CRN) electrophysiological model to simulate the WT, L532P, and N588K mutation conditions. The experimental values for the IKr mutation measured in previous studies were applied to the CRN cell model, and N588K and L532P mutation. The proarrhythmic effect of the KCNH2 ion channel variation was analyzed by comparing the differences in the APD, current density of IKr, wavelength, electrical activation in 2D and 3D geometry, and a vulnerable window between the wildtype (WT) and mutated (MT) genes.
Go to : 
METHODS
To numerically simulate the electrical excitation of the cell, a model of cellular voltage change caused by the flux of ions (Na+, K+, Ca2+, Cl-) in and out of the cell is required. Electrical equilibrium (Hodgkin and Huxley18) is used to describe the changes in intracellular voltage when the bio-electrons entering the cell are injected into or out of the cell.
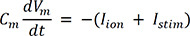
In (1), Cm is cell membrane capacitance per unit cell and Vm is cell voltage. Iion is the total bio-ion current flowing into the cell, and Istim is the amount of perturbation current flowing in and out of the cell to induce excitation of the cell. Iion varies slightly depending on the cell model and channel type.
In this study, we used the CRN (Fig. 1A) model for the human atrium cell model. Similar to the Hodgkin and Huxley model, the CRN modeled the cell membrane as a capacitor connected with variable resistances (ionic channels) and batteries (driving forces) in parallel.19 The ionic channels (Iion) of the CRN model are as follows:
In (2), INa represents rapid inward Na+ current, IK1 is inward rectifier K+ current, Ito is transient outward K+ current, IKur is ultra-rapid delayed rectifier K+ current, IKr is rapid delayed rectifier K+ current, IKs is slow delayed rectifier K+ current, ICa, L is L-type inward Ca2+ current, Ip, Ca is plateau current, INaCa is Na+/Ca2+ exchanger current, and Ib, Na, and Ib, Ca are background Na+ and Ca2+ currents, respectively. Details of this have been described in previous studies.19 We used the CRN model in this study because the mathematical equations of the L532P and N588K mutations were measured and calibrated to be fit with the CRN model. Moreover, it is a well-known electrophysiological model for human atrial cells.
Modified CRN cell model
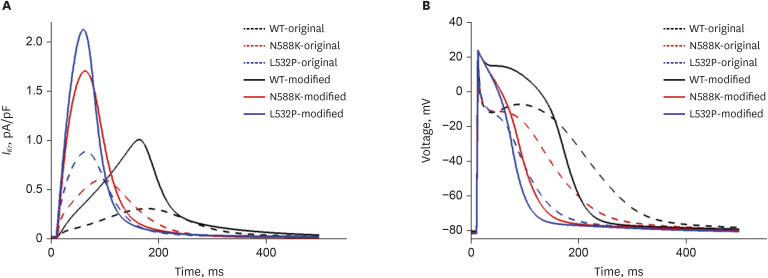
We modified some ion currents to mimic closely related AF phenomena following Jacquemet et al.20 The modified model reduces the Ito, ICa L, and Ikur currents by 80%, 30%, and 90%, respectively, and increases the IKr by 50%, resulting in AF action potential similar to the actual clinical results. We compared the original parameter input of CRN with the modified parameter of that in Jacquemet et al.20 study at the single-cell level (Fig. 2). In the 2D tissue and 3D atrium, we presented the electrophysiological activation based on the modified parameters setting.
Implementation of mutation
The IKr of the CRN model was modified to accommodate IKr changes due to gene mutation (Fig. 1B). For this, maximal IKr conductance, activation, and inactivation gate values, and α- and β-subunit values of the CRN model were substituted into the gene mutation experimental values measured in the previous studies.132122 The L532P mutation was applied to Xenopus laevis oocytes using a double-electrode voltage-clamp assay (Hassel et al.13), and the N588K mutation was examined in Chinese hamster ovary cell counts measured at room temperature, 37°C (McPate et al.21). CRN models using N588K gene mutations in IKr have been described by Loewe et al.22 and assumes the following explanation:
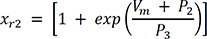
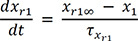
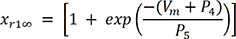
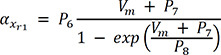
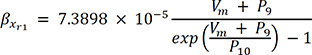
In (3), IKr is rapid delayed outward rectifier K+ current, and xr1 in (4) and xr2 in (5) are the activation and inactivation gating variables, respectively. αxr1 in (7) and βxr1 in (8) are the forward and backward rate constants for the gating variable, and τxr1 in (9) is the time constant for gating variables. P1-11 are experimental values for WT, N588K, and L532P mutation conditions (the values of P1-11 are provided in Table 1).22
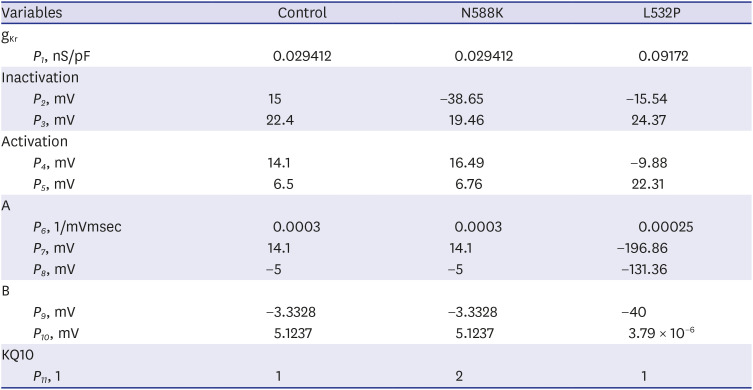
Table 1
Parameter values of the control and mutation condition
The parameters were obtained based on the experimental data from the work of Loewe et al.22
![]()
Tissue model
In 2D models, the spatial resolution was 0.02 cm; this is comparable to the size of atrial cells and provides a stable numerical solution. The size of the 2D atrial sheet model was 10 × 10 cm2. The number of nodes and elements was 250,000 and 249,001, respectively. The purpose of the 2D simulation is to present the electrophysiological activation pattern of both N588K & L532P mutations on a simplified and limited medium. The limited 2D medium can show the arrhythmogenic and the duration of the rotor. The pattern shown can provide some clues of what happens in the 3D heart which is more complex geometry. The mesh of the 3D atrium is shown in Fig. 3. The electrophysiological model of the cardiac tissue was based on the following equation:
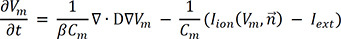
where Vm represents membrane voltage, t represents time, D is the diffusion coefficient of the tissue, Cm indicates membrane capacitance, Iion is the sum of all transmembrane ionic currents, →n is the activation gate variable, and Iext is the externally applied stimulus. The partial differential equation above represents electrical propagation in the cardiac tissue; it covers diffusion in the tissue, and reaction in cardiac channels. The parameter values of the above equations are shown in Table 2.
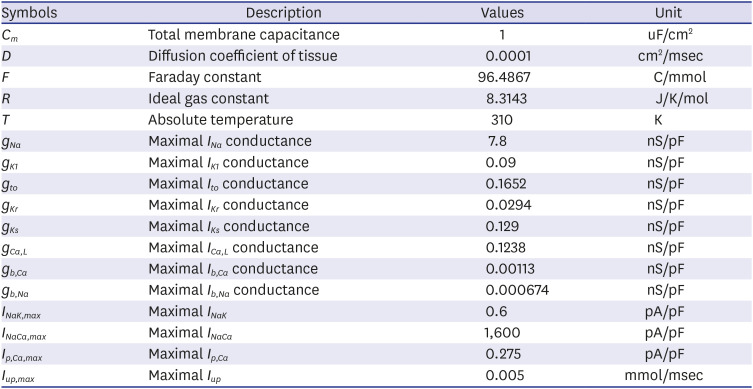
Table 2
Parameter values of the current model
![]()
Computational resource
The hardware that we used for the simulation is a super-computer which consists of 12 processor units divided into two sockets. Each socket has 16 Gb memory so the total memory is 32 Gb with 6 cores in each socket. The processor series is Intel(R) Xeon(R) CPU E5-26400. Each core handles 1 thread per run. The operating system of the computer is CentOS Linux distribution with kernel version 2.6.18-238.3l5. The code to run the simulation is based on the C++ with Portable, Extensible Toolkit for Scientific Computation (PETSc) library for parallel computing.
Simulation protocol
We conducted single-cell simulations of MT and WT conditions with original parameters of CRN and modified maximum conduction values (as mentioned in part A) before performing the 2D and 3D simulation. Single-cell action potential was computed by conditioning the cell models for 30 times, at a basic cycle length (BCL) of 1 seconds with supra-threshold stimuli of 1.5 nA/pF (duration of 2 msec). APD90 was defined as the duration between action potential upstroke and 90% repolarization (−71 mV for CRN model). Diastolic interval (DI) was defined as the time interval between 90% repolarization of the previous action potential and the upstroke of an action potential.
Single atrial cell models have then incorporated into a multicellular 2D sheet and 3D models using simplified homogenous electrophysiology of the human atria. Re-entrant waves in the 2D and 3D models were initiated using a standard S1-S2 protocol. S1-S2 protocol is a common protocol to artificially trigger reentry generation, hence arrhythmia.232425 The reentry occurrence will lead to spiral wave generation (in 2D tissue) and scroll wave (in 3D geometry) depending on the wavelength, conduction velocity, medium, and the effective refractory period. In the 2D model, three S1 stimuli were applied in sequence every 400 msec to produce a planar wave-front propagating in one direction. After allowing four planar waves to pass, a premature stimulus was applied in the lower left quadrant of the patch. Fig. 4A describes the principle of the S1-S2 protocol used in the 2D model (Fig. 4A).
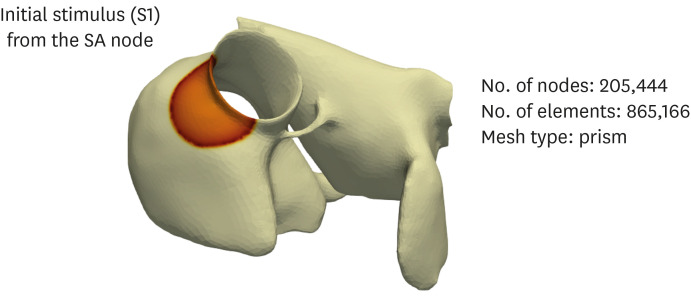
In the 3D model, three S1 stimuli were applied in the SA node (Fig. 4B). When the refractory tail of this wave reached the center of the medium, and S2 stimulus was applied, parallel with the S1 stimulus, but cover only three-quarters of the length of the medium. This produced a second wavefront with a curly tip, generating a spiral wave. Temporal time-lapse was recorded as 0.05 msec for both 2D and 3D simulations.
To reveal the vulnerable time window under MT and WT conditions, we conducted periodic stimulation for all cases by using 3D heart tissue. First, we applied 10-fold of stimuli at the apex to create planar waves from apex to the base with 600 msec cycle length. Then we applied the 11th stimulation periodically in ranged from 450 msec to 150 msec with 10 msec decrease. These methods reveal the vulnerable window which generates reentry, thus arrhythmia, under N588K, L532P, and WT conditions.
Go to :
RESULTS
The comparison of the action potential and IKr current between original parameters of CRN and modified CRN with the WT and MT (N588K, L532P) genes is shown in Fig. 2. The maximum amplitude of IKr current was 1.003, 1.703, and 2.125 (pA/pF) for WT, N588K, and L532P, respectively, and MT showed a greater increase than WT. Also, the current peak time of IKr reached 163.4, 63.6, and 60 (msec) for WT, N588K, and L532P, respectively. For APD90, MT reached its peak value earlier than WT (214.4 msec for WT, 104.99 msec for N588K, 105.26 msec for L532P) with the APD50 of 160.6, 76.8, and 63.2 msec for WT, N588K, and L532P, respectively (Table 3).
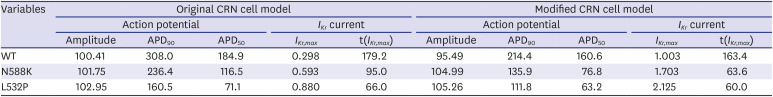
Table 3
Comparison of action potential and IKr current results between the original and modified crn cell models (WT, N588K, L532P)
APD = action potential duration, CRN = Courtemanche-Ramirez-Nattel, IKr = rapid delayed rectifier potassium channel, t = time, WT = wildtype.
![]()
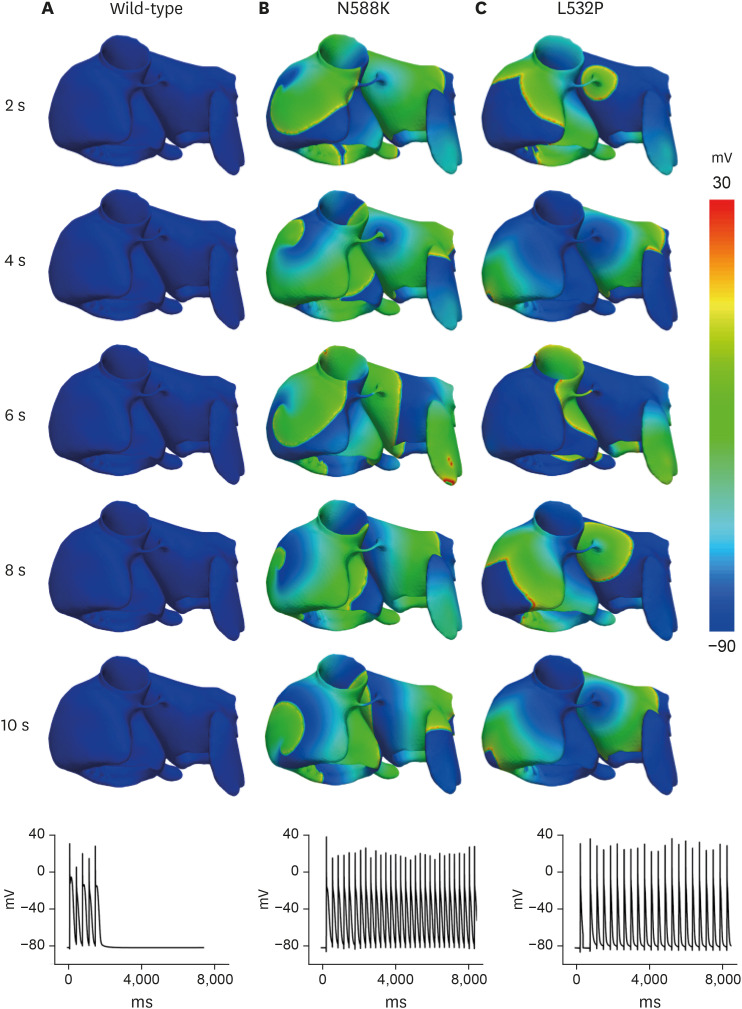
The re-entrant wave pattern of the WT and MT through 2D simulation is presented in Fig. 5. In contrast to self-termination without wave breakup after 2,000 msec from the start of the last S1 stimulus (Fig. 5A), MTs maintained non-termination during the simulation period. The N588K mutation maintained a stable reentrant wave for 10 seconds (Fig. 5B), whereas the L532P mutation had a wave breakup after 3,300 msec from the start of the last S1 stimulus and a fibrillation phenomenon where the waveform split into smaller fragments (Fig. 5C).
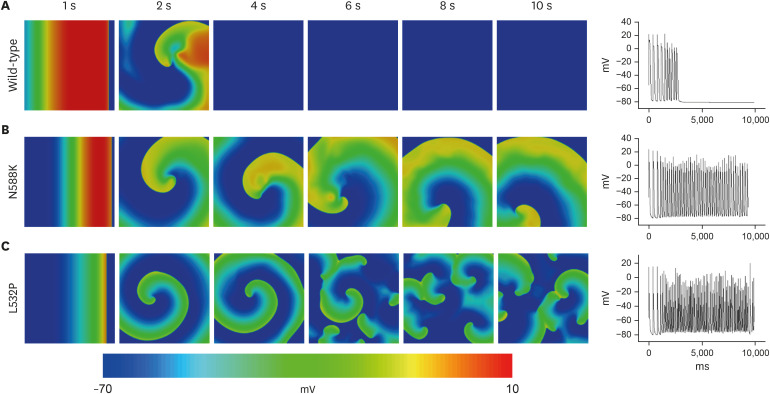
The results of the electrical conduction simulation of the WT and MT for the 3D atrial model are shown in Fig. 6. The wavelengths were in the order of WT ≥ N588K > L532P and the conduction velocities for all three were similar (19.7 cm/sec ± 0.2 cm/sec). As a result of the spiral wave characteristics, the WT was self-terminate at 2 seconds after the S2 stimulus with a rather unstable re-entrant wave (Fig. 6A). In the N588K, a stable re-entrant wave was maintained until the end of the simulation after the S2 stimulus (Fig. 6B). In the L532P, the re-entrant wave progressed and a wave breakup phenomenon occurred generating an irregular waveform (Fig. 6C).
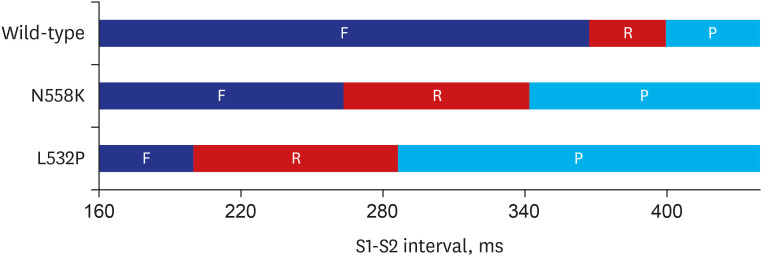
The vulnerable window of the WT and MT according to the 10th stimuli and 11th stimuli interval in the 3D LA model is shown in Fig. 7. The interval of the 10th and 11th stimuli was between 150 msec to 450 msec with a 10 msec resolution. The red block with F indicates the failure to generate an electrical signal in the tissue by the 11th stimulus due to the effective refractory period. The yellow block with R indicates that re-entry occurs by the 11th stimulus within that time interval. The re-entry interval in WT ranged from 360 msec to 400 msec. The re-entry interval in N588K and L532P were 270 msec to 340 msec and 200 msec to 290 msec, respectively. Lastly, the blue block with the letter P indicates a normal planar wave propagation of an electrical signal by the 11th stimulus.
Go to :
DISCUSSION
In this study, we simulated KCNH2 mutations using an electrophysiological 3D atrial model. To this end, we modified two aspects of the CRN cell model. First, we modified the ion currents presented in a previous study and applied them to our cell model.20 Second, the clinical data of N588K and L532P mutations derived from previous studies were assigned to our cell model.1321 This modified cell model for the mutation described by Loewe et al.22 was then applied to the anatomical LA model provided by Yonsei Severance Hospital to observe the KCNH2 mutation phenomenon.
First, the effect of the modified CRN cell model was confirmed by measuring IKr current (Fig. 4A) and AP (Fig. 4B). The maximum amplitude of IKr current in MT conditions was higher than that in the original CRN model, and it was observed earlier than the original CRN model. This change in IKr due to MT conditions resulted in a shortening of the APD, which further resulted in APD50 and APD90 being shorter than that of the CRN model. This confirmed that the modified ion current shortens the APD of the CRN model (Table 3). Shortening of the APD is important to induce fibrillation by increasing the vulnerability of the 2D and 3D models, indicating that the modified CRN cell model can exhibit fibrillation condition realistically.
The application of KCNH2 mutation to the modified cell model confirmed that the N588K and L532P mutations advance the peak IKr current and maximize the amplitude (Table 3). This change in IKr causes early repolarization, shortens the plateau of the AP compared to the normal condition, and ultimately shortens the APD. This suggests a relationship between KCNH2 mutations and the short QT syndrome, and comprehensively explains the increase in tissue susceptibility that maintains and sustains re-entry.
In this study, we analyzed the 2D tissue and 3D LA models by applying the N588K and L532P mutations to the modified cell model and confirmed that vulnerability is increased by the mutation. For the WT, the spiral wave is self-terminated due to the long wavelength that results in the lack of sufficient space for activation in the tissue. However, the N588K and L532P mutations significantly shortened the wavelength, which allows the spiral wave to remain constant. Previous studies have shown that the N588K-hERG SQT1 mutation in 2D and 3D tissues can reduce the minimum substrate size to allow for re-entry.26 These results were confirmed by the N588K mutation effect in this study. In particular, the L532P mutation was found to cause a wave break-up phenomenon after a time-lapse, resulting in irregular conduction-sustaining fibrillation. This confirmed our hypothesis that due to the significant APD shortening by the L532P, the electrical pattern wave propagates in such a chaotic condition resulting in the spiral wave break up (AF condition). The impact of the KCNH2 mutation on the vulnerability of the atrium model is also confirmed by a vulnerability graph in Fig. 7. Re-entrant waves were generated within 40 msec for WT, whereas for the N588K and L532P mutations, they were generated within 80 and 100 msec, respectively. This suggests that the effect of MT increases the vulnerability of the atrium model and that KCNH2 mutation causes AF.
Researchers have reported pharmacological substances that blockade the IKr channel, hence prolonged the QT interval including Vesnarione,27 Macrolide Antibiotic,28 Ketoconazole,29 and Seldane.30 However, none of the studies specifically observed the effect of the IKr blocker on either N588K or L532P mutation conditions. In 2004, Brugada et al.6 attempted to apply Sotalol, IKr blocker, which is categorized as class III antiarrhythmic drugs on N588K-hERG mutation. They found that the N588K mutation decreased the effect of Sotalol on the hERG channel which is consistent with clinical findings. In addition, a proper drug for L532P was open for future pharmacological study.10
The limitations of this study are as follows; First, we used a homogeneous cell model as 2D and 3D heart models, however, in reality, the heart tissues are discrete. Second, we only provide the electrophysiological analysis of the two mutations without considering the mechanical behavior, which previously observed for D172N, G229D, S140G, and V241F mutations.25313233 Third, we were unable to compare our simulation results with experimental data due to the limited availability of patients with N588K or L532P mutations. Nonetheless, this computational study demonstrated the relationship between the L532P and N588K mutations with atrial arrhythmia and AF. This study provides further insights into the N588K and L532P mutations to pharmacologists. Patients with N588K or L532P mutations can benefit from drug therapy with appropriate substance and dosage.34
In summary, we confirmed that the vulnerability of the atria is increased due to KCNH2 gene mutations, and the re-entrant wave is constantly maintained throughout the 3D left atrial model. In particular, the CRN modified cell model confirmed that the L532P gene mutation can cause AF.
Go to :
 XML Download
XML Download