

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Philadelphia-negative (Ph−) classical myeloproliferative neoplasms (MPNs) include polycythemia vera, essential thrombocythemia (ET), and primary myelofibrosis.1 The Ph− MPNs are considered rare diseases, but their incidence rates are constantly increasing.2 According to a recent nationwide population study in Korea, the crude annual incidence per 100,000 of all Ph− MPNs was highest for ET (0.84), followed by polycythemia vera (0.40) and primary myelofibrosis (0.15).2
The majority of the Ph− MPN patients harbor key driver mutations in JAK2, CALR, and MPL, which represent major diagnostic criteria in combination with hematologic and morphological abnormalities in the World Health Organization classification.1 These mutations occur in a mutually exclusive manner in 1 of 3 genes,3 but several reports with co-occurrence of two driver mutations have been reported.4567 Given the paucity of data, clinical characteristics and outcomes for the patients harboring concurrent driver mutations have not been fully evaluated, and their clinical relevance is largely unknown. Here, we present the first case of ET positive for both JAK2 and MPL mutations in Korea. Interestingly, the patient had two independent MPL mutations.
Go to : 
CASE DESCRIPTION
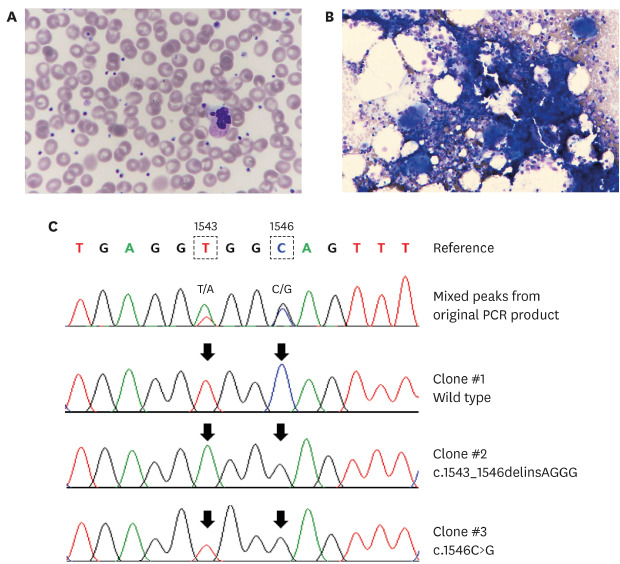
A 57-year-old man visited Soonchunhyang University Bucheon Hospital in October 2019 for evaluation of dizziness. Magnetic resonance imaging showed a tiny dot-like acute infarction in his right cerebellum. His hemogram at diagnosis were as follows: white blood cells, 7.1 × 109/L; hemoglobin, 14.8 g/dL (148 g/L); and platelets, 708 × 109/L. For thrombocytosis, an allele-specific, real-time polymerase chain reaction (PCR) assay (BioSewoom, Seoul, Korea) for JAK2 V617F mutation was performed using his peripheral blood specimen, but the result was negative. About a month later, thrombocytosis was still persistent. Bone marrow aspiration and biopsy were performed to evaluate for the persistent thrombocytosis. The peripheral blood smear and bone marrow biopsy revealed a marked increase in platelet and megakaryocyte numbers without evidence of dysplasia (Fig. 1A and B). He had no organomegaly. No history of inflammatory or infectious disorders, hemorrhage, or other causes of reactive thrombocytosis was found.
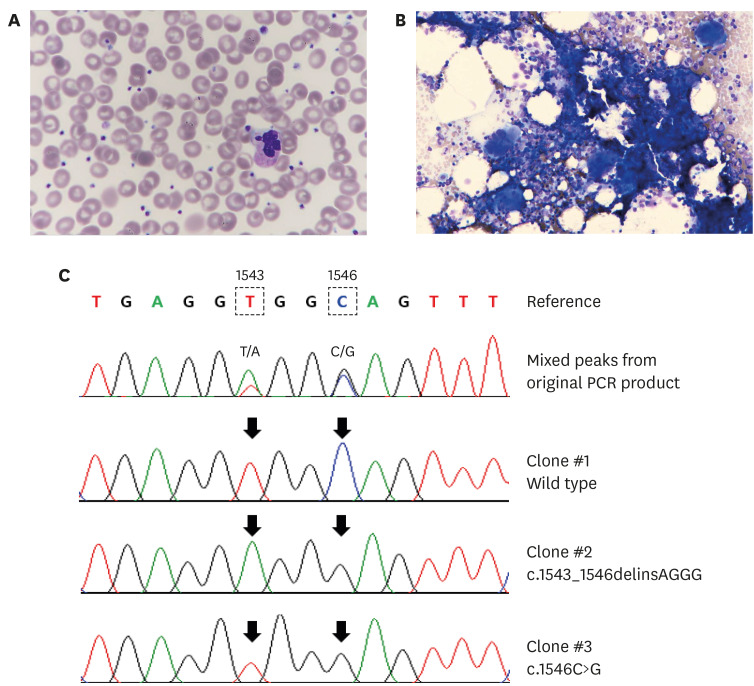 | Fig. 1Hematological and molecular characteristics of an essential thrombocythemia patient with coexisting JAK2 and MPL driver mutations. (A) Peripheral blood is unremarkable, except for thrombocytosis (708 ×109/L) (Wright-Giemsa stain, ×400). (B) Bone marrow aspirate exhibits normal cellularity but increased number of large megakaryocytes with hyperlobulated nuclei (hematoxylind and eosin stain, ×400). (C) Segments of the MPL sequence electropherogram of the original PCR product, and of three TA-clones obtained from a second PCR product revealed wild-type, c.1543_1546delinsAGGG, and c.1546C>G in the mix. Note the correspondence between the variable sites (arrows) distinguishing the cloned sequences and the double peaks observed in the original sequence.PCR = polymerase chain reaction.
|
Genetic analysis was conducted with informed consent from the patient. The karyotype was normal. We studied mutations of JAK2 V617F, CALR exon 9, and MPL exon 10 in the patient using samples obtained from bone marrow aspiration. An allele-specific, real-time PCR assay (BioSewoom) was used to detect the JAK2 V617F mutation. The CALR, MPL mutations were assessed by Sanger sequencing (primer sequence and reaction conditions are available upon request). Unlike the previous result on peripheral blood, the JAK2 V617F mutation study was positive on bone marrow aspirates. Subsequently, the mutation allele burden was calculated as the percentage of the V617F copy number to the sum of V617F and wild-type copy number by quantitative real-time PCR (Biosewoom); it was 4% (1,270/32,970). The patient was negative for the CALR mutation. However, atypical mutations in MPL were found, with two substitutions that changed from thymidine to adenine and cytosine to guanine at nucleotides 1543 and 1546 near each other, respectively (Fig. 1C). To determine whether these two substitutions were located in the same clone, we performed a TA-clone experiment and detected multiple clones of MPL mutations. The dominant clone was c.1543_1546delinsAGGG (p.Trp515_Gln516delinsArgGlu) observed at a frequency of 78% (14/18 clones). In addition, the same patient harbored c.1546C>G (p.Gln516Glu) mutation at a frequency of 17% (3/18). The wild-type sequence was only detected in 6% (1/18). These three distinct sequences were located on independent colonies, indicating the presence of three different clones.
Based on the above results, he was finally diagnosed with ET. Treatment began with aspirin and hydroxyurea. He was discharged from the hospital after improvement and was instructed to continue regular hospital visits and routine blood evaluation.
Ethics statement
This case was approved by the Institutional Review Board of the Soonchunhyang University Bucheon Hospital (SCHBC 2019-12-007) and exempted from informed consent.
Go to :
DISCUSSION
Coexistence of two driver mutations in Ph− MPN is a rare event. A prior report of 685 CALR mutated MPN patients estimated that 0.6% and 0.3% of its subjects had concurrent JAK2 and MPL mutations, respectively.8 Several previous studies on the mutational spectrum of JAK2, CALR and MPL in Korean patients with MPN did not find patients with concurrent mutations.910 However, these rates may be an underestimation of co-occurrence frequencies. Another domestic study, by Kang et al.,6 reported a higher rate in their evaluation of coexistence of JAK2 and CALR mutations (4%, 7/167 ET patients), although no coexistence with MPL mutation was found. They analyzed that the difference in reporting rates between groups may be due to differences in the sensitivity of the test method to detect small amounts of mutant allele burden.6 We summarized previously published ET cases with co-occurrence of two driver mutations in Table 1. Although the coexistence effect of driver mutations on Ph− MPN is not clearly known, a previous report of 40 double mutant patients with ET showed a man predominance, advanced age at onset, low hemoglobin, and high platelet count, but the occurrence of thrombosis during follow up was not different compared to ET patients with a single mutation.4 Our patient is man and had acute cerebellar infarction, but it is difficult to estimate the long term prognosis yet due to his short follow-up period.
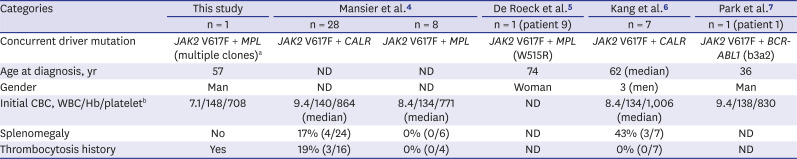
Table 1
Summary of patients with essential thrombocythemia carrying coexisting driver mutations
| Categories | This study | Mansier et al.4 | De Roeck et al.5 | Kang et al.6 | Park et al.7 | |
|---|---|---|---|---|---|---|
| n = 1 | n = 28 | n = 8 | n = 1 (patient 9) | n = 7 | n = 1 (patient 1) | |
| Concurrent driver mutation | JAK2 V617F + MPL (multiple clones)a | JAK2 V617F + CALR | JAK2 V617F + MPL | JAK2 V617F + MPL (W515R) | JAK2 V617F + CALR | JAK2 V617F + BCR-ABL1 (b3a2) |
| Age at diagnosis, yr | 57 | ND | ND | 74 | 62 (median) | 36 |
| Gender | Man | ND | ND | Woman | 3 (men) | Man |
| Initial CBC, WBC/Hb/plateletb | 7.1/148/708 | 9.4/140/864 (median) | 8.4/134/771 (median) | ND | 8.4/134/1,006 (median) | 9.4/138/830 |
| Splenomegaly | No | 17% (4/24) | 0% (0/6) | ND | 43% (3/7) | ND |
| Thrombocytosis history | Yes | 19% (3/16) | 0% (0/4) | ND | 0% (0/7) | ND |
CBC = complete blood count, WBC = white blood cell, ND = no data, Hb = hemoglobin.
ac.1543_1546delinsAGGG (p.Trp515_Gln516delinsArgGlu) and c.1546C>G (p.Gln516Glu); bValues are presented in the International System of Units (WBC, ×109/L; Hb, g/L; platelet, ×109/L).
![]()
It is of note that the JAK2 V617F allele burden in our patient is very low (4%) compared to the relatively high (17%–78%) allele frequency of MPL mutations. Mansier et al.4 reported that second concurrent mutations are more frequently encountered in patients with JAK2 V617F mutant allele frequencies of < 5% like our patient. The low JAK2 mutant burden might be explained by preexisting clonal hematopoiesis before overt signs of MPNs,11 followed by the acquisition of a second oncogenic mutation of CALR or MPL leading to the MPN phenotype.4
Gain-of-function mutations in exon 10 of the MPL lead to cytokine-independent activation of JAK2 and STAT protein signaling.12
MPL p.Trp515Leu (W515L) or p.Trp515Lys (W515K) is the most frequent mutant type, but several other substitutions have also been reported, including p.Trp515Arg (W515R), p.Trp515Ala (W515A), and p.Ser505Asn (S505N).12 In the current study, we identified two different clones of MPL mutations, the dominant clone being c.1543_1546delinsAGGG (p.Trp515_Gln516delinsArgGlu) and the smaller clone being c.1546C>G (p.Gln516Glu). A recent in vitro cell-based assay showed that these two MPL mutations identified in this study are gain-of-function mutations inducing cytokine-independent growth advantages.13 We suggested that the activating MPL mutations may be disease-causing factors participating in the pathogenesis of ET. The presence of multiple mutations within the same gene is rarely observed, and the molecular mechanism causing the molecular complexity and their clinical impacts are still unclear. We believe that this report will contribute to a better understanding of the genetic and clinical features of Ph− MPN patients with concurrent driver mutations.
Go to :
 XML Download
XML Download