

 PDF
PDF Citation
Citation Print
Print


Introduction
Diamond-Blackfan anemia (DBA) usually presents within the first year of life with normochromic and usually macrocytic anemia. DBA is a heterogeneous genetic disorder characterized by a defect in erythroid stem cells, congenital anomalies, growth retardation and cancer predisposition [1-3].
Chronic red blood cell (RBC) transfusions, corticosteroid therapy and hematopoietic stem cell transplantation (HSCT) are the mainstays of therapy [1-7]. Some patients are steroid-responsive, whereas others remain transfusion-dependent and may develop iron overload leading to chelation therapy [8]. HSCT can be an option if corticosteroid treatment is not successful or causes unacceptable side effects.
As a result of iron overload, the endocrine glands may be impaired in DBA patients treated with transfusions [2,5,9]. Iron overload-related endocrine complications include adrenal insufficiency, hypothyroidism, growth hormone (GH) deficiency, diabetes mellitus, diabetes insipidus, and hypogonadism [2]. Increased awareness can help clinicians diagnose endocrinopathies and promptly treat it to improve the quality of life for DBA patients [10].
Here, we present the case of an 18-year-old boy with DBA and iron overload-associated endocrinopathies. Moreover, we provide an overview of the current state of knowledge as well as recommendations for the day-to-day care of patients with DBA [10].
Go to : 
Case
The patient was an 18-year-old boy diagnosed with DBA and had been receiving chronic RBC transfusions. He initially came to the hospital for poor eating and dyspnea during lactation at 3 months of age. Complete blood count presented with severe normocytic anemia and bone marrow biopsy showed nearly absent erythroid precursors compatible with diagnosis of DBA syndrome. At the time of diagnosis, he was given blood transfusions and then started on corticosteroid therapy but showed no response. We considered allogeneic HSCT but matched donor was not found so he received RBC transfusions every 3 weeks thereafter at our pediatric oncology department. He also received iron chelation therapy with deferoxamine and deferasirox. He was referred to the pediatric endocrine department for evaluation of endocrine abnormalities after hyperglycemia was detected.
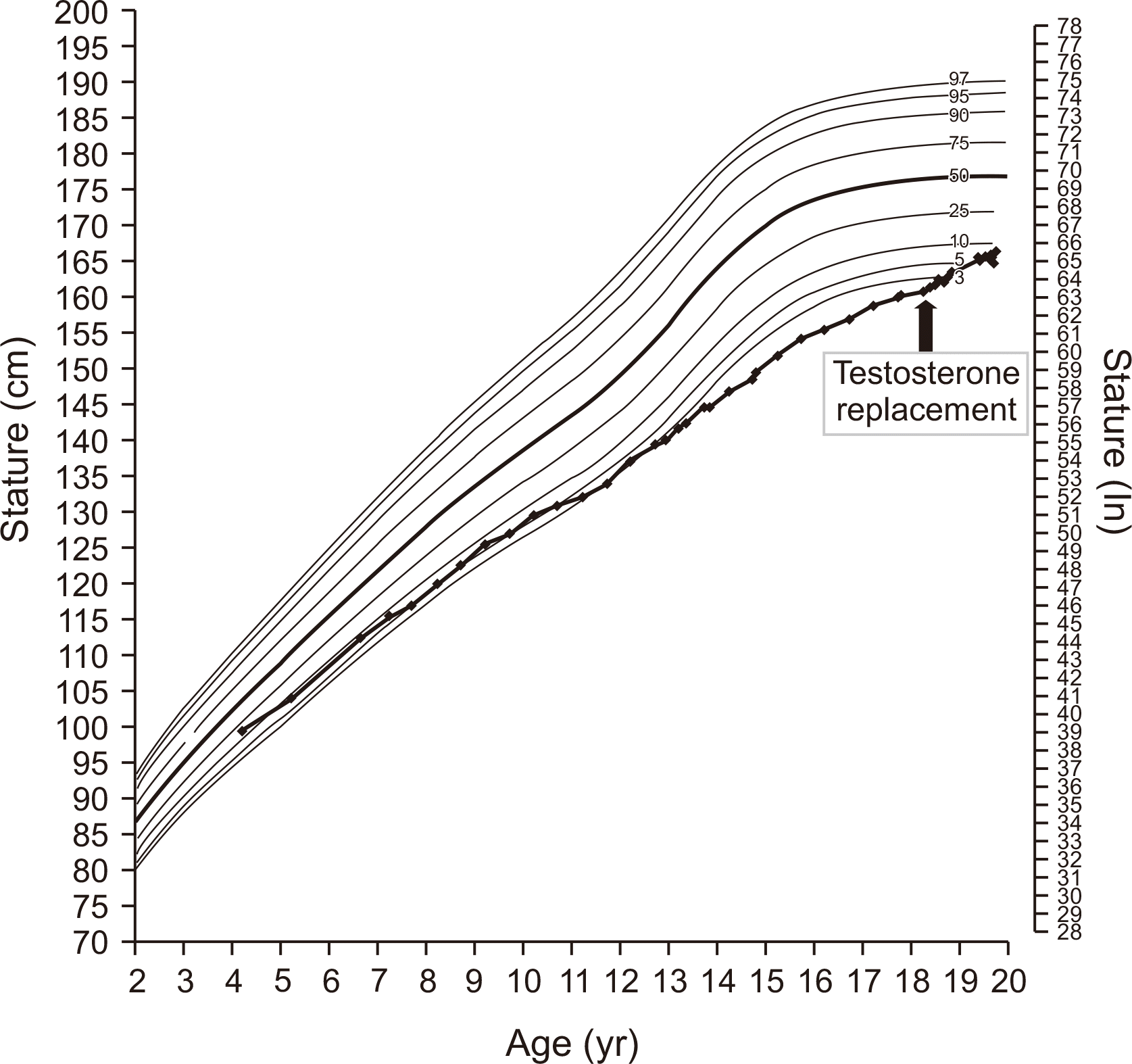
When he was referred, physical examination was conducted; his height, weight, and body mass index were 160.3 cm (<3rd percentile), 64.7 kg (25th–50th percentile) and 25.18 kg/m2 (>95th percentile), respectively. Mid-parental height was 173.0 cm. Growth velocity was below 4 cm/yr (Fig. 1). He exhibited a lack of secondary pubertal characteristics with small testes (right 1.5 cc and left 2.0 cc), a sparse beard and sparse pubic hair, and no change in voice; however, gynecomastia was not present. Secondary sexual maturity was classified as Tanner stage 1.
Laboratory blood tests revealed anemia (hemoglobin 6.4 g/dL, hematocrit 17.8%), normal liver function (aspartate aminotransferase/alanine aminotransferase 26/39 IU/L), a high triglyceride level (290 mg/dL), a low-normal high-density lipoprotein–cholesterol (49 mg/dL), a high fasting insulin level (32.5 μIU/mL), fasting hyperglycemia (207 mg/dL), and a high glycated hemoglobin (HbA1c) level (7.8%). Serum ferritin levels were high with a range of 1,141–1,584 ng/mL (reference range, 30–400 ng/mL).
Results of endocrine function tests showed a low insulin-like growth factor-1 (IGF-1) level (118.67 ng/mL), a low testosterone level (0.16 ng/mL), a normal basal luteinizing hormone (LH) level (1.3 mIU/mL), a normal basal follicle stimulating hormone (FSH) level (4.4 mIU/mL), and 25-OH vitamin D deficiency (9.4 ng/mL). Thyroid function was normal (Table 1). In the gonadotropin-releasing hormone stimulation test, the peak LH level was 5.9 mIU/L and the peak FSH level was 6.7 mIU/L (Table 2). The amplitudes of LH and FSH responses were low for his bone age (15 years).
Table 1
The patient’s laboratory results of metabolic components and endocrine hormone
AST, aspartate aminotransferase; ALT, alanine aminotransferase; γ-GTP, γ-glutamyl transpeptidase; ACTH, adrenocorticotropic hormone; DHEA-sulfate, dehydroepiandrosterone sulfate; ADH, antidiuretic hormone; LH, luteinizing hormone; FSH, follicle stimulating hormone; IGF-1, insulin-like growth factor-1; TSH, thyroid-stimulating hormone; T3, triiodothyronine; fT4, free thyroxine; PTH, parathyroid hormone.
![]()
His bone age was markedly delayed by 3 years compared to chronological age (Fig. 2A). We performed pituitary magnetic resonance imaging (MRI), which revealed no physiologic hyperplasia of the pituitary gland. A total fatty marrow change involving the skull and the upper C-spine was observed, likely due to DBA. (Fig. 2B). He was placed on echocardiogram and electrocardiogram monitoring. The findings were normal. He had short stature, delayed bone age, and decreased IGF-1 levels and was expected to have a GH deficiency. His mother, however, refused GH stimulation test and GH replacement.
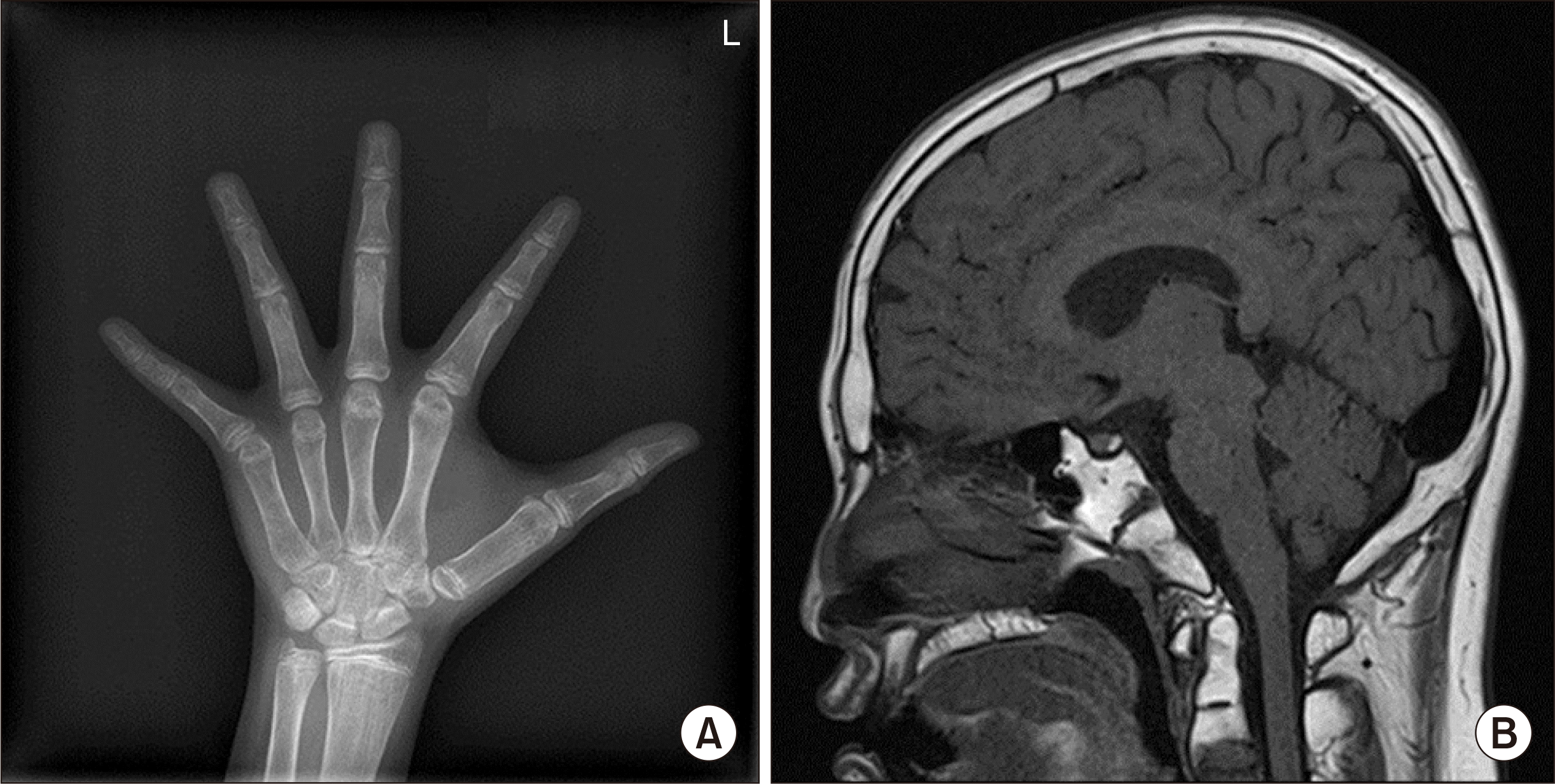 | Fig. 2(A) Anterior-posterior view of the left hand: the bone age was 15 years (chronological age, 18 years). (B) Pituitary magnetic resonance imaging: pituitary magnetic resonance imaging showing lack of physiologic hyperplasia of the pituitary gland, but no evidence of abnormal enhancement or mass-like lesion. A total fatty marrow change involving the skull and upper C-spine is noted, probable due to Diamond Blackfan anemia. Patient’s consent for photo publication was obtained.
|
He was diagnosed with hypogonadotropic hypogonadism, and he received testosterone replacement therapy (testosterone enanthate 50 mg intramuscular, slow dose escalation up to 200 mg). He was educated on the importance of exercise and diet to control blood glucose levels for diabetes mellitus. He was advised to take oral vitamin D supplements (cholecalciferol 1,000 IU per day).
After 1 year of testosterone replacement therapy, testicular volume increased (right 6 cc and left 6 cc), and growth velocity also increased (Fig. 1). In addition, HbA1c level was monitored every 6 months and improved from 7.8% to 7.1% after 1 year and a half.
This case report was approved by the institutional review board of Ewha Womans University Seoul Hospital (2019-12-002-001). Patient, himself, gave written consent for publication.
Go to :
Discussion
In early infancy, patients with DBA, a rare, inherited bone marrow failure syndrome, develop congenital (erythroid) hypoplastic anemia. It is diagnosed based on signs and symptoms such as pale skin, irritability, sleepiness, heart murmur, and low birth weight and based on the results of blood and bone marrow testing. A DBA diagnosis is confirmed based on the following four results from complete blood count and bone marrow biopsy: anemia seen prior to turning age one, normocytic or macrocytic RBCs, a low reticulocyte count (number of new RBCs), and normal bone marrow sample with only a few cells that will develop into RBCs. DBA is characterized by RBC disorders, congenital anomalies, and a predisposition to cancer [1-3]. These patients are often associated with short stature with a multifactorial etiology.
There are three therapeutic approaches regarded as the mainstays of DBA treatment: RBC transfusions, corticosteroids and HSCT, of which the latter is used mostly as a rescue therapy [1]. HSCT from a compatible donor can help restore the bone marrow’s ability to make RBCs, and may be tried if steroid therapy is not successful. Corticosteroid treatment is the only treatment option proven to be of use for ineffective erythropoiesis in DBA. While an initial course of corticosteroid treatment shows a response in 80% of patients, the definite mechanism of its positive clinical effect is yet verified [1]. The other 20% that do not respond require chronic RBC transfusion therapy [10]. RBC transfusions are an effective option to treat anemia in DBA due to the fact that bone marrow dysfunction in DBA usually is solely related to erythrocyte production. However, the toxicity associated with iron overload, accompanying chronic transfusion treatment, is a limiting factor for long-term transfusions [1]. Recurrent transfusions often result in iron overload, which leads to chelation therapy [8].
Endocrine dysfunction is common in patients with DBA, and symptoms are observed even at a young age [1,10]. Over 50% of patients with DBA have one or multiple endocrinopathies [1,2,8]. Clearly, long-term treatment with corticosteroids and iron overload following chronic blood transfusions play an important role in the development of endocrine dysfunctions, with symptoms including impaired growth, adrenal insufficiency, osteoporosis, diabetes and pubertal delay [1,2,5,8,9]. In addition, iron overload is associated with hypogonadism and hypothyroidism [1]. Given that some endocrine abnormalities reflect significant and potentially irreversible organ damage, it is advised to regularly screen for endocrine dysfunction.
In this case, the patient initially had failed corticosteroid therapy and considered allogeneic HSCT but a matched donor was not found. Eventually, he received constant RBC transfusions every 3 weeks for a long time, which led to symptom manifestations of hypopituitarism (growth impairment and hypogonadism) and diabetes mellitus. The patient was thought to have acquired hypopituitarism caused by iron overload, which resulted in hypogonadism and short stature. The patient showed normal levels of other pituitary hormones such as adrenocorticotropic hormone, thyroid-stimulating hormone, prolactin, and antidiuretic hormone (vasopressin). Chronic RBC transfusions may cause endocrine disorders, among which diabetes is the most common problem caused by insulin resistance and impaired insulin secretion. Testosterone replacement therapy induced secondary sexual characteristics (increased testicle size and increased pubic hair) and increased growth velocity in our patient. He controlled his high blood glucose levels through exercise and dietary control instead of depending on drugs.
Despite the use of iron chelation agents from the early stages of the disease, the patient still showed iron overload. Several factors could affect the effectiveness of chelation therapy including the availability of a given chelator and its properties, drug tolerability, transfusional iron burden and the patient’s compliance.
The goal of iron chelation therapy is to reduce the body burden of iron. The specific aim is to minimize the production of reactive oxygen species, thus reducing damage to critical organs such as the liver, heart, and endocrine organs, resulting in reduced morbidity and improved survival. The guidelines for iron chelation therapy agree that patients with DBA receiving RBC transfusions are the most likely to benefit from the treatment. Iron chelation must be given over extended periods of time and must be rigorously followed.
A major limitation of this case report is that routine pituitary MRI performed to confirm pituitary hypoplasia in this patient could not be used to detect iron deposits. As a result, radiologic correlation between iron burden and the presence of endocrinopathies could not confirmed. However, MRI of pituitary gland is not routinely used to diagnose iron overload. Furthermore, ferritin is a marker that reflects the amount of iron stored in the body and represents a predictive factor for the progression of endocrine dysfunctions. His pituitary MRI finding such as absent physiologic hyperplasia of the pituitary gland showed indirect evidence of iron overload.
While there were significant improvements in our understanding of the pathophysiology of DBA, there were limited advances in DBA therapy [11]. Although endocrinopathies have been reported in patients with DBA, there are no specific endocrine guidelines for patients with DBA that can be utilized for screening. Regular assessments of growth (height and weight) and puberty are recommended. It is important for patients with iron overload to undergo precise evaluation and long-term follow-up care to determine the incidence of hypopituitarism. In patients with reduced growth velocity or short stature; bone age and indicators of GH deficiency (IGF-1 and IGF-B3) should be analyzed. Hormone replacement therapy is crucial in improving the quality of life and clinical outcomes of patients with hypopituitarism [2]. Specifically, GH therapy should be considered as per indication, and patients should be evaluated for delayed puberty. Monitoring should continue into adulthood for detecting secondary gonadal failure. Patients receiving chronic corticosteroid therapy or those at risk for iron overload should be monitored for glucose intolerance in addition to being screened for adrenal and pituitary dysfunction. An endocrine work-up can reveal the presence of diabetes mellitus with a minimum insulin secretory response. Annual screening for diabetes mellitus should be performed by evaluating HbA1c levels or using an oral glucose tolerance test [1]. In addition, it is recommended to prescribe vitamin D supplementation in all DBA patients and perform periodic bone mineral density measurements for osteoporosis. It is advised that patients aged >14 years, particularly those who received transfusion, screen for hypothyroidism. In sum, with regards to the complexity of potential endocrine dysfunction, it is advised that an endocrinologist should be involved in the management of all chronically transfused patients and children that are treated with steroids for more than 1 year.
This report highlights specific features of a patient with endocrine dysfunction associated with DBA who was treated with chronic transfusions. DBA is a rare disease, and therefore, the incidence of various complications associated with it is also low. Sample cases of treating such condition are not common. In this case report, he received transfusion treatment according to existing regimen and appropriate chelation, but eventually developed endocrinopathy. We can conclude from this that patients of any age with normal results on screening laboratory examinations should not be excluded from long-term monitoring, since endocrinopathies may develop later [1]. We reiterate the need for awareness and close monitoring of various complications that may lead to iron overload from repeated transfusions. Our case report emphasizes the importance of periodic, meticulous evaluation of endocrine function in patients with transfusion-associated iron overload. Timely diagnosis and management may help avoid further morbidities [1].
Go to :
 XML Download
XML Download