

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Next-generation sequencing (NGS), also known as massively parallel sequencing or high-throughput sequencing, is a technology allowing simultaneous sequencing of millions of DNA or RNA sequences. The advantages of NGS compared with traditional sequencing methods include higher throughput with sample multiplexing, higher sensitivity in detecting low-frequency variants, faster turnaround time for high sample volumes, and lower cost. NGS represents a true sequencing technology revolution after Sanger sequencing [1]. Sequencing the first human genome using Sanger sequencing required many years and billions of dollars; however, with the emergence of NGS, a complete human genome can now be sequenced within a few days for less than $1,000 [2]. NGS has a wide spectrum of applications in laboratory medicine and has become an integrated part of precision medicine. The technology has been widely used in diagnosis, prognosis, and therapy selection for constitutional disorders, oncology, and infectious diseases [3–5]. Concurrently, an increasing amount of well curated clinical, genetic, and genomic data is being generated by NGS, further driving the development of precision medicine [6]. The U.S. Food and Drug Administration (FDA) recently released a set of guidelines for the design, development, and validation of NGS tests and approved several NGS-based tests and targeted therapies [7–9]. In addition, the Centers for Medicare & Medicaid Services (CMS) has been actively monitoring the rapid innovation of NGS tests and working to ensure coverage of NGS-based tests. All these advances have accelerated the clinical application of NGS in laboratory medicine. This review highlights recent developments in NGS technologies and their clinical application in diagnosis, prognosis, and therapeutics of inherited diseases, cancers, and infectious diseases.
Go to : 
NGS TECHNOLOGIES
Historically, DNA sequencing technologies have played important roles in molecular biology and clinical fields [10–13]. The first-generation platform, Sanger sequencing, was developed by Fred Sanger in 1977 and has been used for decades in research and clinical genetics [14–16]. Three decades later, NGS technologies have evolved rapidly, leading to the invention of second and third generation sequencing technologies. Sequencing turnaround time and cost have been dramatically reduced since 2001 when the first draft map of the human genome was accomplished [17–26]. In this section, we discuss the commonly used next generation sequencers and their strengths and challenges.
Second generation sequencing technologies
Various second-generation sequencing technologies have been developed by different commercial companies. Overall, the workflows of the different sequencing technologies include three steps: (1) template preparation including nucleic acid extraction; (2) library preparation including clonal amplification; and (3) sequencing and alignment of short reads.
Roche 454 sequencing (Roche, Basel, Switzerland), launched in 2005, was the first commercially available massive parallel sequencing platform. Roche 454 sequencing uses pyrosequencing technology, which captures pyrophosphate (PPi) release and uses it as an indicator of specific base incorporation. Fragmented DNA is bound to beads with ligated adaptors followed by fragment amplification via emulsion PCR within an emulsion droplet [27]. The beads containing multiple copies of the same DNA template are then loaded into PicoTiterPlate (PTP) wells. Each nucleotide is sequentially flowed into the PTP wells. Each time a nucleotide is incorporated during DNA synthesis, it releases pyrophosphate, which is converted to ATP. In the presence of ATP, luciferase coverts luciferin to oxyluciferin to generate light, which is then detected and captured by a coupled-charge device (CCD) camera [28–30]. Sequencing accuracy is dependent on the reading of the light signals. A misread or missing signal, especially in homopolymer sequencing, could result in base errors and insertions or deletions. The Roche 454 sequencer genome sequencing (GS)-FLX, launched in 2008, could generate approximately 700 Mb of sequence data per run with read lengths up to 1,000 bases in approximately 20 hours [31]. Roche 454 was phased out of the NGS field in 2016 because of its much higher cost compared with other high throughput NGS sequencers such as the Ion Torrent (Thermo Fisher, Waltham, MA, USA) and Illumina (San Diego, CA, USA) systems.
Unlike other technologies using fluorescence or chemiluminescence, Ion Torrent uses sequencing via hydrogen ion detection technology, which detects the release of protons while nucleotides are being incorporated into the strands during synthesis. The fragmented DNA is attached to 3-micron diameter beads with specific adapter sequences. Clonal amplification happens via emulsion PCR on the beads [27], and the beads are then loaded into microwells. The change in pH due to proton release, generated by the incorporation of each base during synthesis, is detected by the sensing layer of the microwell, which translates the chemical signal into a digital one [32]. The first Ion Torrent Personal Genome Machine (PGM) sequencer was released in 2010. PGM has an output of up to 2 GB per run and fast run time (2–7 hours), which is suitable for targeted sequencing or smaller/genomes [32, 33]. In 2012, Ion Torrent released the Proton sequencer, which provides a higher throughput at the same speed and is capable of sequencing both exomes and human genomes [34, 35]. Compared with the PGM and Proton sequencers, the Ion GeneStudio S5 series sequencers, launched in 2015, changed the instrument cartridges and reagents resulting in easier preparation and shorter run time [36]. Rather than relying on the laser scanners/CCD cameras used in other sequencing technologies, the Ion Torrent platform is more rapid, direct, and less expensive. However, Ion sequencers do have sequencing error issues such as artifact insertions/deletions (indels) associated with homopolymeric stretches and repeats [35].
Illumina platforms are currently widely used in the NGS field [37–43]. Illumina developed a bridge PCR approach for clonal amplification and sequencing by reversible termination technology. Both ends of the fragmented DNA anneal to two fixed adapters, which are immobilized to the solid surface of the flow-cell, followed by bridge amplification to form clusters that contain clonal DNA fragments. Each reversible terminator (RT) nucleotide (ddATP, ddGTP, ddCTP, ddTTP) is protected at the 3′-OH group and contains a cleavable fluorescent dye. Modified RT nucleotides are incorporated into the growing DNA chains during synthesis and release fluorescent signals, which are captured and recorded using a CCD camera. This technology significantly reduces the homopolymer sequencing error by incorporating a single base at a time, as the addition of another base requires that a terminator first be removed [44, 45].
MiSeq (Illumina), one of the most prevalent benchtop sequencers, was launched in 2011. It can produce data ranging from 540 Mb to 15 Gb, which is suitable for sequencing small panels of genes and bacterial genomes [46]. The production-scale sequencer, HiSeq2500 (Illumina), was launched in 2012, with the capacity of sequencing an entire genome in approximately 24 hours. Both platforms use the four-channel sequencing by synthesis (SBS) system, in which each base is detected by individual images. NextSeq 500, launched in 2014, uses two-channel SBS system, which only requires two images to determine all four base calls. This new technology reduces imaging capture time and the number of cycles and hence, decreases sequencing cost and time. The HiSeq X Ten, HiSeq 3000, and HiSeq 4000 systems were launched in 2015; these adopted billions of pre-formatted nanowell grids at fixed locations rather than normal flow-cells, resulting in many folds higher data output compared with MiSeq and HiSeq2500 [45]. NovaSeq, the most powerful sequencer to date, was released in 2017, with the goal of reducing the cost of sequencing a human genome to $100. NovaSeq also uses two-channel chemistry, but with larger flow-cells with more nanowells and a faster imaging capture system. It can generate up to 6 Tb of sequence data and 20 billion reads in approximately two days. NovaSeq allows customers to choose from four different flow cell types with different capacities to meet different sequencing needs. Overall, Illumina platforms are currently the most popular in both clinical and research settings owing to their high accuracy, relative low cost, and high throughput.
Third generation sequencing technologies
Although the second-generation sequencing technologies have hugely impacted the NGS field, we still face many challenges such as short sequence reads leading to sequence gaps, alignment issues associated with repetitive regions or pseudogenes, and PCR artifacts. To overcome these limitations, third generation sequencing technologies, single molecule sequencing-based technologies, were developed [47–49]. Pacific Biosciences (Menlo Park, CA, USA) (Pac Bio) single molecule real-time (SMRT) and Oxford Nanopore sequencing technologies (Oxford Nanopore Technologies, Oxford, UK) are representatives of this generation [50].
PacBio SMRT technology does not require amplification and offers much longer reads than second generation sequencing technologies. The library preparation is similar to that of second-generation sequencing technologies, except that the adapters used in library preparation have a hairpin structure to ensure that the double-stranded DNA fragments become circular after ligation to form the SMRTbell template. The bases are sequenced by synthesis in real time on a chip containing millions of zero mode waveguides (ZMWs), which are nanowells several nanometers in diameter and approximately 100 nm in depth. The template molecule and DNA polymerase are immobilized at the bottom of each ZMW. During the sequencing reactions, the complementary strand of the template is elongated by DNA polymerase with fluorescently labeled deoxyribonucleotide triphosphates. The CCD camera inside of the machine captures and records the fluorescent signals in real-time observation [48, 50, 51]. The first PacBio RS (PacBio) was released in 2011, with an average sequencing read length of approximately 1.5 kb [49, 52]. Two years later, RS II was released, with an average read length of approximately 20 kb [48, 53]. In 2015, PacBio launched a new SMRT system, Sequel, with larger cells and an increased number of ZMWs, which produces an average read length between 8–12 kb [53]. The upgraded Sequel II system can currently generate eight-fold the sequence data with 50% of the reads ≥50 kb. PacBio technology has a few advantages compared with the second-generation sequencing technologies, including much shorter sample preparation time (4–6 hours) and sequencing run time (within a day/run), much longer sequencing reads (average 10–15 kb), and reduced GC bias and sequencing errors due to PCR amplification. However, this technology has a few inherent drawbacks including a relatively high error rate (10–15%). Most errors are due to indels, and a small portion is caused by miscalls. This error rate can be reduced by multiple sequencing runs [53, 54].
The other representative third-generation sequencing technology is Oxford Nanopore; this novel technology uses nanopores, tiny biopores with a nanoscale diameter, and measures current changes, instead of SBS or fluorescence detection approaches. Any particle passing through the pore interrupts the voltage across the channel owing to the nature of the nanopore; each of the four bases leads to a distinctive current change owing to their unique structures. No amplification step or fluorescence labeling is required by this platform, and it does not rely on DNA polymerase like PacBio SMRT. Not only can Nanopore sequence DNA, it can also directly sequence RNA and protein [48]. This technology also has the advantages of short turnaround time and no GC bias. The most apparent disadvantage of the Nanopore technology is a high sequencing error rate of approximately 14%, and most of the errors are indels.
Overall, the third-generation sequencing technologies provide longer sequence reads, which helps close gaps in current reference assemblies generated from short reads and can sequence through extended repetitive regions and characterize structural change in human genomes. However, third generation technologies still have a major issue of high error rate. A hybrid sequencing strategy, combining second- and third-generation NGS technologies, could address some of these issues [55–59]. The background technology, read length, and sequencing capacity of commonly used NGS platforms are summarized in Table 1.
Table 1
Summary of commonly used NGS platforms
![]()
Go to :
APPLICATION OF NGS IN CONSTITUTIONAL DISORDERS
The evolution of technologies for the diagnosis of constitutional disorders
NGS is rapidly transforming how research into the genetic determinants of constitutional disorders is performed. The technique is highly efficient, with detailed genetic information produced in a reasonably short time and at a relatively low cost. Several studies have compared the diagnostic yield and cost of NGS with other types of DNA testing. G banding, for example, detects chromosomal aberrations, with a diagnostic yield of approximately 3% for unexplained constitutional disorders [60]. High-resolution chromosomal microarray analysis (CMA) detects gene copy number variations (CNVs) and has a diagnostic yield of 15–20% for the same disease categories [60]. NGS whole-exome sequencing (WES) has a diagnostic yield of 25% for Mendelian disorders [61] and whole-genome sequencing (WGS) has a slightly higher diagnostic yield (27%) for pediatric and adult genetic diseases [62]. In contrast to WES and WGS, targeted NGS gene panels focus on subsets of genes, ranging from several to hundreds, depending on the focus of the specific diseases. The diagnostic yields of NGS panels vary significantly; for example, congenital glycosylation disorder panel has a reported diagnostic yield of 14.8% [63], while the prenatal skeletal dysplasia panel has a diagnostic yield of 53% [64]. In this section, we focus on the process from translational research to clinical diagnosis, molecular diagnosis rate, and patient care, as well as NGS methods in constitutional disorders.
Forward and reverse phenotyping
Genetic disease studies traditionally progress from phenotype to genotype analysis, the so-called “forward genetics” method [65]. NGS has led to a new process known as reverse phenotyping, in which the genetic marker data are used to drive, or form the basis of, new phenotype definitions [66]. The combination of NGS and segregation analysis may identify a pathogenic variant in a gene that was not known to cause the disease or was previously linked to a different phenotype. In such cases, retrospective clinical interpretation of the patient and their family members can reveal additional characteristics that were unrecognized previously. In a review of over 300 WES studies investigating the causes of rare diseases (between 2010–2012), 178 studies reported a novel disease-associated gene, 51 discussed reverse phenotyping, and 79 reported novel or known variants in a known disease-associated gene [67]. More recent studies have shown that approximately 25% of reported variants in known disease-associated genes are associated with a phenotype that matched the clinical presentation of the investigated patient [68–71]. These findings highlight the advantages of the “genotype-first” approach in studying rare diseases, especially when phenotypic presentations vary drastically from one patient to another. Thus, NGS provides a powerful molecular tool for establishing a clinical diagnosis even before the disease characteristics are fully revealed.
The number of newly identified disease-associated genes has grown exponentially since the application of NGS technology. The identification of a new disease-associated gene or the association of a known gene with new phenotypes is of great significance for patient management including early therapeutic intervention in some cases [72]. The most commonly used approaches for identifying disease-associated genes include: (1) analysis of a group of patients with the same clinical characteristics using WES or WGS and filtering out rare variants in a common gene in all or some members of the group; and (2) proband analysis in conjunction with parents or informative family members and filtering out variants by disease inheritance pattern (autosomal dominant, recessive, X-linked, or de novo) to simplify the process and identify the causal gene.
NGS Strategies for the diagnosis of constitutional diseases
Targeted NGS gene panels
Targeted NGS gene panels are designed for a specific disease or group of diseases, with the ability to maximize coverage, sensitivity, and specificity for the genes of interest. Therefore, targeted NGS gene panels often have higher diagnostic yield than exome sequencing (ES) or genome sequencing (GS). However, when diagnostic uncertainty is high, the diagnostic rate of a targeted NGS gene panel can be lower [73].
Targeted NGS gene panels are often used in the context of a suspected disease or a group of diseases. Diagnostic rates vary across NGS gene panels. For instance, a study using a genetic eye disease panel, including 257 genes, the mitochondrial genome, and previously identified deep intronic pathogenic variants in a cohort of 192 probands, identified a causal variant in 98 of the probands, with a diagnostic rate of 51% [74]. The diagnostic rate for genetically heterogeneous diseases, such as hereditary hearing loss, could be increased by using a combined approach. A study using tiered ES reported a diagnostic rate of 21% with Tier 1 testing including Sanger sequencing and targeted deletion analysis of the two most common nonsyndromic hearing loss genes (STRC and GJB2) and two mitochondrial genes (MT-RNR1 and MT-TS1). The diagnostic rate increased to 33% with Tier 2 testing, including 119 genes and CNV analysis, using array comparative genomic hybridization [75].
The cost of targeted NGS gene panels is variable, but usually significantly lower than that of ES. More expensive panels often incorporate multiple sequencing and copy number analysis techniques to improve the sensitivity of the test.
ES
A study of 500 unselected consecutive patients who received traditional genetic diagnostic evaluations showed that nearly half of the patients remained undiagnosed [76]. Clinical ES targets approximately 22,000 protein-coding genes, increasing the chances of identifying pathogenic variants that may be causal for genetic diseases. ES can also identify risk variants for a specific condition/syndrome that have not been diagnosed in the individual tested; these results are called secondary or incidental depending on whether the gene is deliberately studied. Guidelines for the clinical reporting of these categories of findings have been published [77].
Clinical ES is regularly used for patients with previous negative NGS gene panel tests or complex phenotypes with broad differential diagnoses. This approach has the advantage of evaluating all known disease-associated genes and collecting sequencing data for future reanalysis, as variant classification and new gene discovery advances. When ES is used in patients with a suspected genetic disease without a diagnosis, the molecular diagnostic rate ranges from 24 to 52% [78–80].
Many studies have aimed to improve diagnostic yield; for example, including the biological parents (trio testing) together with proband testing. In one study, the molecular diagnostic rate increased by 16% by confirming the de novo status of a variant in the proband or clarifying segregation patterns for recessive diseases by identifying the variant in both unaffected parents [81]. Trio testing with clinical phenotyping strengthens diagnosis confidence. Previous studies have also reported an increased diagnostic rate using re-analysis; a 15% improvement was achieved by periodical reanalysis of genotype/phenotype data of undiagnosed individuals [82].
GS
GS is a comprehensive method for analyzing entire genomes. A comparison between GS and ES with six unrelated individuals demonstrated that an estimated 650 high quality coding single nucleotide variants (SNVs; approximately 3% of coding variants) were detected by GS but missed by ES [83]. Studies comparing GS with CMA followed by a targeted NGS gene panel, the standard of care for first-tier clinical investigation of congenital malformations and neurodevelopmental disorders, showed that GS identified clinically diagnostic genetic variants in 34% cases, which was more than a two-fold increase compared with CMA plus a targeted NGS gene panel (13%) [84]. For inherited retinal diseases, GS identified 14 clinically relevant genetic variants in 46 individuals; these variants included large deletions and variants in noncoding regions of the genome. These findings confirmed a molecular diagnosis for 11 of 33 individuals referred for GS who had not obtained a molecular diagnosis through targeted NGS gene panels, suggesting GS could result in an overall 29% increase in diagnostic yield [85].
More recently, rapid GS (rGS) has been used for infants with acute illness. rGS enables the identification of potential causes of a genetic disease or ruling out a genetic etiology for a condition within a period of 36 hrs. During a nine-month period, infants from 42 families underwent rGS for etiologic diagnosis of genetic diseases, with a diagnostic yield of 43% (18 of 42 infants). Twenty-six percent (11/42) of the infants who underwent diagnostic rGS avoided morbidity, one had a 43% reduction in likelihood of mortality, and one started palliative care. In six of the 11 infants, the changes in management because of the rGS results reduced inpatient cost by $800,000–2,000,000 [86]. rGS provides a faster diagnosis, enabling timely precision medical interventions that can decrease the morbidity and mortality of infants with genetic diseases [87].
A meta-analysis on the cost-effectiveness of ES and GS based on 36 studies showed that a single test ranged from $555 to 5,169 for ES and from $1,906 to 24,810 for GS. Most cost-efficiency studies have concluded that ES and GS are economically superior to other testing options [88].
In summary, targeted NGS gene panels contain a set of genes specifically designed for a (or a group of) known genetic disease and can detect mosaic variants with higher confidence owing to higher sequencing depth. The diagnostic yield is relatively higher for patients with typical clinical characteristics. However, targeted panels can only detect variants in genes included in the panel and therefore are not suitable for patients with uncharacteristic manifestations and limit approaches for new discovery such as reverse genotyping. ES includes nearly all protein-coding genes, and GS covers nearly the entire genome, providing much higher diagnostic yield, but also increased cost at present.
Clinical impacts
A study examining the application of NGS for 83 patients with suspected inherited bone marrow failure syndrome demonstrated that a causal variant was detected in 18% of the patients [98]. In 20% of these patients, the results led to the initiation of a cancer surveillance program and proper family counseling [89]. In another study involving 278 infants referred for ES, 36.7% received a genetic diagnosis, and the medical management was affected in 52.0% of the diagnosed patients [90].
Apart from the diagnostic value, the ultimate value of any diagnostic test is its impact on patient treatment, which is dependent on knowing when to order the test and whether therapeutic choices exist.
Go to :
APPLICATION OF NGS IN ONCOLOGY
Cancer is a genomic disease, and the identification of characteristic genomic aberrations in cancers has become an integral part of precision medicine. NGS can be used to identify different genomic alterations commonly observed in cancer including SNVs, small indels, CNVs, and fusion genes in hematologic or solid malignancies [91–93]. Although the availability of whole genome, exome, or transcriptome sequencing has been increasing, targeted gene sequencing is the method of choice in clinical laboratories for cancer diagnosis to ensure optimal sequencing quality (read depth and coverage, variant characterization, reporting), cost-effectiveness, and turnaround time. Small NGS panels (<50 genes) can be applied for specific cancers such as acute myeloid leukemia (AML) or breast cancer; however, larger NGS panels are commonly used in academic hospitals and commercial laboratories for a wide range of cancers. In our clinical laboratory in the Division of Genomic Diagnostics (DGD) at the Children’s Hospital of Philadelphia (CHOP), large custom-designed NGS panels have been developed for hematologic malignancies and solid tumors for the detection of SNVs/indels, CNVs, and fusions (Fig. 1) [91, 94]. When we tested these custom-designed NGS panels on 367 pediatric cancer samples, we found that NGS panel testing had a clinical impact in 88.7% of leukemia/lymphomas, 90.6% of central nervous system (CNS) tumors, and 62.6% of non-CNS solid tumors in the cohort [91]. NGS application in clinical laboratories is regulated by a number of agencies such as the FDA, Centers for Disease Control and Prevention (CDC), and other agencies [7, 95]. The standards and guidelines for the validation of NGS panels, validation of bioinformatics pipelines, and interpretation and reporting of sequence variants in cancers were recently published [96–98].
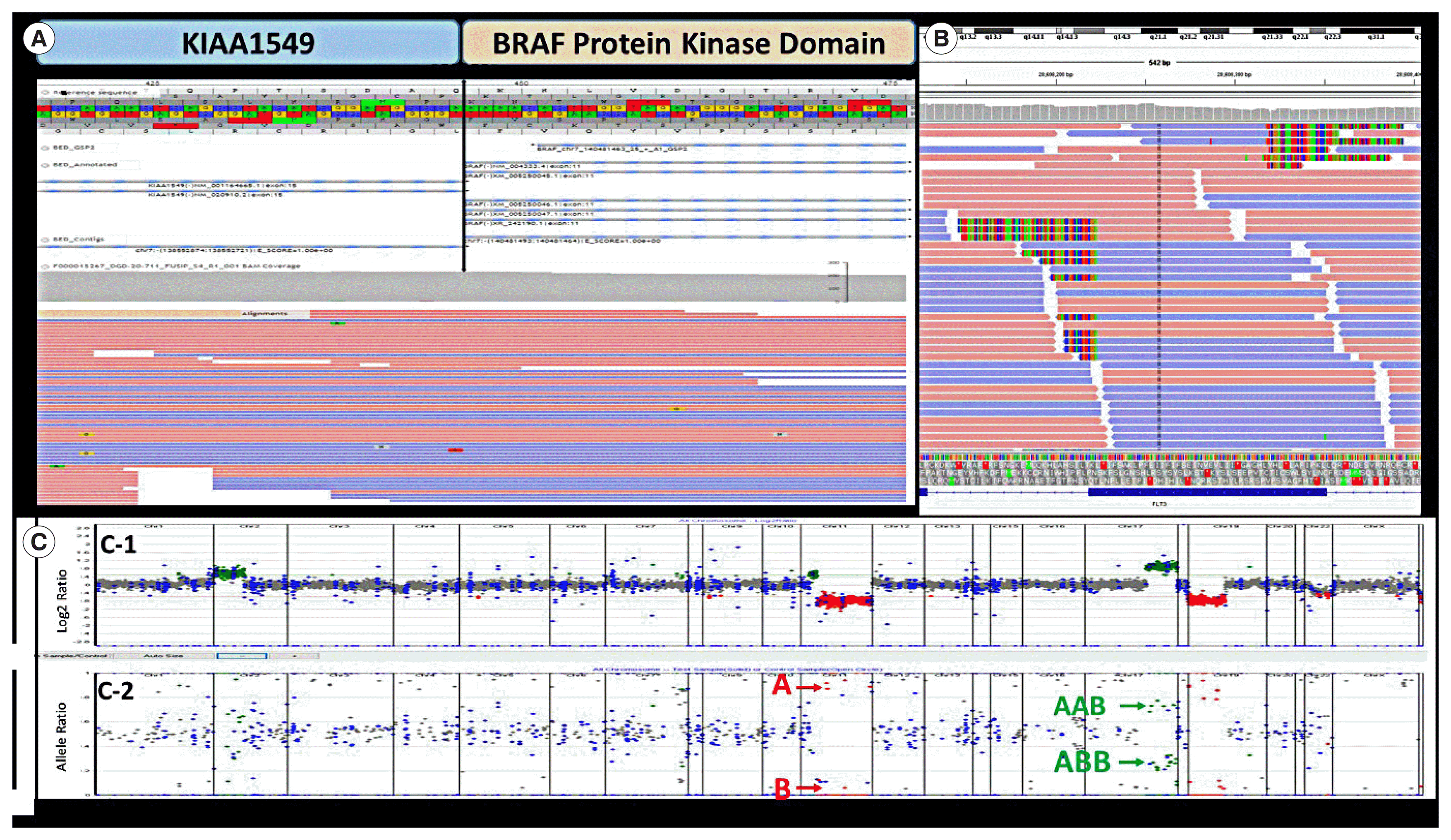 | Fig. 1Representations of genomic alterations identified by the Children’s Hospital of Philadelphia Division of Genomic Diagnostics NGS tests. (A) A KIAA1549-BRAF fusion detected in the Fusion panel. Red and blue represent forward and reverse sequencing reads. (B) FLT3-ITD identified in the NGS solid tumor panel. (C) CNVs identified by the NGS solid tumor panel. (C-1) CNV analysis based on reading depth. (C-2) B allele frequency analysis demonstrating SNV separation. Red arrows indicate one copy of 11q; green arrows indicate three copies of the 17q genomic region.
Abbreviations: NGS, next-generation sequencing; CNV, copy number variation; SNV, single-nucleotide variation.
|
FDA-approved NGS panels in cancer
The Thermo Fisher (Waltham, MA, USA) Oncomine Dx Target Test was the first companion diagnostic (CDx) test approved by the US FDA in June 2017. This test simultaneously evaluates variants in 23 genes associated with non-small cell lung cancer (NSCLC). The approved markers include BRAF V600E, EGFR L858R, EGFR exon 19 deletions, and ROS1 fusions for use as a CDx for specific targeted therapies [99]. Thermo Fisher is actively expanding the Oncomine Dx Target Test by validating other variants such as ERBB2 exon 20 insertions and other fusions including ALK and RET [100]. The Oncomine Dx Target Test has received positive reimbursement decisions from the CMS and from large commercial health insurers in USA. It has been adopted for use by several national reference laboratories.
The US FDA approved Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT) panel, developed by the Memorial Sloan Kettering Cancer Center, and the FoundationOne CDx (F1CDx), developed by Foundation Medicine, for in vitro diagnosis via detection of genetic variants in November 2017 [101–103]. The MSK-IMPACT test detects SNVs, indels, and microsatellite instability (MSI) in 468 unique genes by sequencing both tumor samples and matched normal samples from the same patient with solid malignant neoplasms to detect genetic alterations that might help guide treatment options. F1CDx detects SNVs, indels, limited CNVs, and gene rearrangements in 324 genes and two genomic signatures, including MSI and tumor mutational burden (TMB), in any solid tumor to identify patients who may benefit from treatment with targeted therapies. Additionally, F1CDx was approved by the US FDA in 2019 to be used as a CDx for olaparib for first line maintenance therapy in BRCA-mutated advanced ovarian cancer [104]. The F1CDx test has also been accepted by the CMS. MSK-IMPACT and F1CDx are single-site tests performed at the Memorial Sloan Kettering Cancer Center and Foundation Medicine, respectively.
The NantHealth Omics Core test was approved by the US FDA in November 2019. It is a WES platform to report TMB and somatic alterations (SNVs and indels) in 468 cancer-relevant genes (https://www.accessdata.fda.gov/cdrh_docs/reviews/K190661.pdf). The Illumina TruSight Oncology 500 (TSO 500) test was launched in October 2018. It targets 523 genes for SNV and indel detection and 55 genes for fusion and splice variant detection. It can also detect immunotherapy-associated biomarkers such as TMB and MSI; Illumina is seeking US FDA approval of this test as a CDx. MI Transcriptome CDx (Caris Life Sciences, Irving, Texas, USA) is an NGS test using RNA from formalin-fixed paraffin embedded tumor tissue to detect structural rearrangements and measure gene expression in cancer patients; it has received a breakthrough device designation from the US FDA for the detection of FGFR biomarkers including gene fusions in solid tumors (https://fdahealthnews.com/stories/512479753).
Application of NGS in cancer diagnosis, prognosis, and therapeutics
Disease-specific biomarkers
In addition to US FDA-approved NGS panels, many other commercial or custom-designed NGS panels are being used in clinical laboratories, with an increasing number of identified biomarkers for cancer diagnosis, prognosis, and therapeutics. For example, in normal karyotype AML, the most frequently mutated genes include NPM1, CEBPA, FLT3, DNMT3A, TET2, IDH1, IDH2, NRAS, ASXL1, KMT2A, WT1, and RUNX1 [105]. NPM1 is the most frequently mutated gene in adult type AML, and NPM1 mutations are associated with favorable outcomes in the absence of concurrent DNTM3A and FLT3-ITD mutations [106]. CEBPA mutations are commonly bi-allelic and have been associated with a favorable outcome in AML [107]. RUNX1 mutations have been associated with worse overall survival [108]. Patients with FLT3-ITD mutations often have an increased risk of relapse and shorter overall survival compared with patients without the mutation [109].
In 2017, the US FDA approved midostaurin, a multi-targeted protein kinase inhibitor, for treating newly diagnosed adult AML patients with FLT3 mutations. In 2017, the US FDA also approved enasidenib for the treatment of relapsed or refractory adult AML with an IDH2 gene mutation, while in 2018 it approved ivosidenib as the first treatment for adult patients with relapsed/refractory AML and an IDH1 mutation [110]. DNMT3A mutations tend to be associated with shortened overall survival [111], and TET2 mutations are often associated with a poor prognosis in cytogenetically normal AML [112–114]; however, patients with TET2 mutations may respond to hypomethylating agents [115]. Mutations in ASXL1, KMT2A, or TP53 are generally associated with poor prognosis [116]. The 2017 World Health Organization (WHO) classification recognizes AML with mutated NPM1, CEBPA, and RUNX1 as specific entities [117].
Another successful example of clinical NGS application is in lung cancer. Certain mutations in several genes, such as EGFR, ALK, and ROS1, are targetable driver mutations [118–120]. There are several US FDA-approved EGFR inhibitors, such as afatinib, gefitinib, erlotinib, and osimertinib, for NSCLC patients with these mutations. Lung cancer patients with ALK rearrangements (e.g. EML4-ALK) may benefit from US FDA-approved ALK-inhibitors, including crizotinib, alectinib, and ceritinib [121]. ROS1 mutations are present in 1–2% of NSCLC patients who may be sensitive to crizotinib and entrectinib, approved by the US FDA in 2016 and 2019, respectively [122, 123]. According to the 2018 guidelines of the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology, testing for mutations in EGFR, ALK, and ROS1 is required for all advanced-stage lung cancers; if these are negative, additional targetable alterations in BRAF, MET, RET, ERBB2, and KRAS can be tested [124]. The use of NGS technology in lung cancer tests has proven reliable, cost-effective, and efficient [92].
Cancer type-agnostic biomarkers
In addition to disease-specific biomarkers, a few pan-cancer biomarkers have been identified, including MSI, TMB, and neurotrophic tropomyosin-related kinase (NTRK). MSI is caused by the inactivation of the DNA mismatch repair (MMR) system and has been found in many types of primary cancers [125, 126]. In May 2017, the US FDA approved the PD-1 inhibitor pembrolizumab (Keytruda) for treating adult and pediatric patients with unresectable or metastatic solid tumors that have been identified as MSI-high (MSI-H) or MMR deficient (dMMR) [127]. This was the first time that a cancer treatment was approved on the basis of a common biomarker, irrespective of the tissue of origin. Furthermore, the US FDA approved other drugs, including nivolumab (Opdivo) and combination of nivolumab and ipilimumab (Yervoy, a cytotoxic T-lymphocyte-associated protein 4 (CTLA-4 inhibitor)), for treating adult and pediatric patients with MSI-H or dMMR metastatic colorectal cancer [128, 129].
NTRK is the second US FDA-approved tissue-agnostic biomarker for cancer treatment, which is based on the presence of NTRK fusions. Fusions involving members of the NTRK oncogene family, NTRK1/NTRK2/NTRK3, are the most prevalent in rare adult cancer types and a broad range of pediatric cancers, with increased frequency in highly aggressive cancers such as glioblastoma [130, 131]. In November 2018, the US FDA approved larotrectinib (VITRAKVI), an oral and selective tropomyosin receptor kinase (TRK) inhibitor, for treating adult and pediatric patients with metastatic or unresectable solid tumors that have an NTRK fusion without a known acquired resistance mutation [132, 133]. In August 2019, the US FDA granted approval for another TRK inhibitor, entrectinib (ROZLYTREK), for adults and pediatric patients (≥12 years of age) with metastatic or unresectable solid tumors that have an NTRK fusion without a known acquired resistance mutation. Entrectinib was also approved for adults with metastatic NSCLC whose tumors are ROS1-positive [134, 135].
TMB is another tissue-agnostic marker for potential response to immunotherapy. TMB can be used to identify patients most likely to benefit from immunotherapy across a wide range of cancer types [136, 137]. TMB can be accurately measured through NGS targeting a large panel of gene (e.g., F1CDx), ES (e.g. NantHealth Omics Core), or GS. Calculation of TMB and identification of MSI status can be simultaneously performed in a single NGS-based test.
Clinical trials
Another application of NGS in oncology is identifying and enrolling patients into the appropriate clinical trials. There are two kinds of clinical trials: umbrella trials and bucket trials (also known as basket trials) [138, 139]. An umbrella trial enrolls patients with a type of morphologically defined cancer and assigns them to a treatment arm on the basis of the genetic mutations detected in their cancer; many different treatment arms exist under the umbrella of a single trial. In a basket trial, patients with different cancers, but sharing the same genetic abnormality, are enrolled to test a new drug against the common genetic alteration. An example of a basket trial is the ongoing NCI-MATCH (National Cancer Institute-Molecular Analysis for Therapy Choice) trial launched in 2015 by the US NCI [140]. Thousands of cancer biopsies from patients undergo NGS to identify genetic abnormalities that may respond to selected targeted therapies.
Application of NGS in minimal residual disease (MRD) monitoring and liquid biopsy
MRD refers to the small number of cancer cells that remain in the body during or after cancer treatment. MRD can be used to measure the effectiveness of treatment, predict the risk of relapse, confirm or monitor remission, and potentially identify an early relapse of the cancer [141]. The most widely used tests for measuring MRD include flow cytometry, real-time quantitative PCR, digital PCR, and NGS. Flow cytometry or PCR-based tests can usually measure MRD down to 1 in 10,000 or 1 in 100,000 cells [142, 143]. The ClonoSEQ test, developed by Adaptive Biotechnologies (Seattle, WA, USA), is an NGS-based test for assessing and monitoring MRD in patients with multiple myeloma and B-cell acute lymphoblastic leukemia. It was approved by the US FDA in 2018 and has been covered by Medicare and private health insurers since 2019 [144, 145]. This test detects as few as one cancer cell in one million healthy (<10−6) cells. The company is also validating this test for other disorders, such as chronic lymphocytic leukemia and non-Hodgkin’s lymphoma. Recently, the US FDA issued draft guidelines on the use of MRD assessment in trials involving patients with hematologic malignancies [6].
Cancer heterogeneity, limited cancer biopsy samples, and invasive procedures are some of the challenges in molecular diagnostics and disease monitoring of solid tumors [146]. Liquid biopsy, which tests circulating cancer cells, circulating cell-free cancer DNA/RNA, or exosomes in blood or other bodily fluids, such as urine and cerebrospinal fluid (CSF), shows great promise for MRD detection, real-time monitoring of disease progression, therapy selection, and cancer diagnosis during early stages of the disease [147–150]. In 2016, the US FDA approved the Cobas EGFR Mutation Test v2, a PCR-based liquid biopsy test from Roche, for detecting specific EGFR mutations in blood samples. This test can be used to identify patients with metastatic NSCLC who would be eligible for treatment with erlotinib (Tarceva) [151]. Several large NGS-based liquid biopsy tests are available, including Guardant360 (Guardant Health) and FoundationOne Liquid (Foundation Medicine); however, these have yet to be approved by the US FDA [152]. The FDA has granted a breakthrough device designation for several blood-based cancer tests, such as the Grail multi-cancer blood test for early cancer detection [153] and the PapGene test for detecting ovarian and pancreatic cancers based on a combination of circulating cancer DNA (ctDNA) and protein biomarkers [154]. Other investigational liquid biopsy tests include TruSight Oncology 500 ctDNA (Illumina), Oncomine Pan-Cancer Cell-Free Assay (Thermo Fisher), and CancerSEEK (Thrive Earlier Detection Corp, Cambridge, MA, USA) [155].
According to the joint review issued by the American Society of Clinical Oncology and the College of American Pathologists, more evidence is needed to demonstrate the clinical utility of liquid biopsy [156]. Additional challenges need to be addressed before a wide application of liquid biopsy in clinical settings. For example, the fraction of cancer-derived DNA in blood plasma samples is generally low; therefore, modified sample preparation methods and much deeper sequence coverage are needed to achieve sufficient sensitivity. Furthermore, clonal hematopoiesis may potentially confound the results of liquid biopsy tests [157].
Go to :
CLINICAL APPLICATIONS FOR DIAGNOSIS OF INFECTIOUS DISEASES
Globally, infectious diseases remain one of the most significant overall causes of morbidity and mortality [158]. Proper and accurate diagnosis of a pathogen is critical for patient treatment, as a delayed or incorrect diagnosis can lead to a multitude of adverse events including unnecessary use or misuse of antibiotics, increased healthcare costs, and worsened patient outcomes [159–162]. Many of these issues are exacerbated in settings, such as poor and under-served areas, in which tools for rapid, accurate diagnoses are limited [163].
Sequencing bacterial DNA and RNA has been used for decades to identify casual pathogens and resistance genes in clinical isolates and even before the onset of NGS, could yield rapid results with high specificity [164, 165]. However, with the advance of NGS technologies, clinicians and laboratory professionals have seen tremendous growth and opportunity for using sequencing as a front-line diagnostic tool. Below, we summarize and briefly highlight the current applications of NGS for infectious diseases including a brief comparison of NGS methods, epidemiological surveillance, identification of pathogens and their resistance markers for diagnosis and treatment, and the importance of the host microbiome.
NGS strategies for infectious diseases
Before discussing the utility of NGS in clinical care of infectious disease, it is important to understand the three main types of NGS methods and how they differ. Targeted NGS uses panels of known pathogen sequences to screen clinical isolates. The panels can be specific for or target multiple types of pathogens including bacteria [166], viruses [167], and even eukaryotic organisms [168]. These panels can also target pathogens known to be involved in particular illnesses, such as gastrointestinal [168] or respiratory [169] diseases, and have been optimized for use with specific sample types, such as CSF [170]. The advantages of these panels are their high specificity, sensitivity, rapid turnaround time, and ability to sequence directly from a host isolate [166, 168, 170]. However, the downsides include their limited scope and inability to identify novel pathogens or antibiotic resistance markers.
In the case of bacterial samples, WGS enables the sequencing of an entire pathogen genome including plasmids. This broad sequencing allows for the identification of antibiotic resistance profiles, which can be used to inform first-line drug use decisions [171]. The drawback of WGS for bacterial samples is that it usually requires a separate culture step to ensure the sample is free of contaminant or commensal bacteria; however, sequencing directly from a host isolate and skipping culture has been performed with targeted enrichment [172]. Furthermore, while WGS datasets accurately define known drug resistance markers, the discovery of novel mutations and their effects on phenotype bring added uncertainty to the test [173, 174].
Metagenomic NGS (mNGS) can use samples directly obtained from a patient and amplify the sequences of all organisms in the sample, including host sequences. This unbiased approach allows for the detection of multiple types of pathogens in one sample (and even the host response to them) and can be particularly useful when targeted or less comprehensive tests are not diagnostic [175–177]. Furthermore, mNGS can detect pathogen sequences that comprise a very small portion of the overall sequenced reads; such low-level sequences can easily be missed by other methodologies [178]. However, there are significant drawbacks to using an mNGS approach, which include the cost and complexity of the process, as well as the need for optimization and standardization of each step in the test, from sample preparation to data analysis [179–181]. Owing to its unbiased nature, mNGS requires additional considerations such as the low ratio of pathogen to host DNA or RNA; unless the host genome or transcriptome are also being analyzed, the host DNA/RNA should be removed [182]. In addition, the presence of commensal bacteria in host samples [183] and contaminated laboratory reagents [184–186] can also confound testing, leading to incorrect results.
Epidemiology and public health
During infectious disease outbreaks, it is critical to rapidly track the transmission, spread, and evolution of pathogens; NGS has been playing an increasingly critical role in these processes. One case that highlights the potential speed of NGS in the field is the West Africa Ebola virus (EBV) outbreak of 2015. Researchers utilized third generation Nanopore technology (as described in the NGS Technologies section above) to track EBV transmission of separate viral lineages across countries by monitoring mutation rates [187]. Using a MinION sequencer (Oxford Nanopore Technologies), researchers were able to sequence over 140 EBV samples directly in the field [187]. Sequencing on the machine itself generally took less than one hr and with cloud-based computing, data were ready to be analyzed in less than a day. Only days after the outbreak of new coronavirus associated pneumonia in Wuhan, China, the sequence of the new coronavirus, SARS-CoV-2, was determined using mNGS, which facilitated disease diagnosis and surveillance, and the development of effective drugs for disease treatment and vaccines for prevention [188].
NGS has also facilitated disease outbreak tracking in clinical settings, particularly in cases of healthcare-acquired infections. In one study, the spread of adenovirus in a neonatal intensive care unit was linked to eye examinations, based on confirmation that the exact viral sequence was found in both affected patients and the equipment used during their examinations [189]. In another hospital, an outbreak of a fungal bloodstream infection in 18 patients receiving anti-nausea medication was found to be connected to contaminated medication, with all patients and contaminated containers testing positive for an identical pathogen identified by WGS [190]. In another case of unexpected transmission, researchers proposed a reasonable hypothesis for the spread of a highly resistant Klebsiella pneumonia strain based on WGS, which linked all cases to an index patient discharged three weeks before any others demonstrated symptoms [191]. NGS can also provide critical insights into the diversity of clinical isolates and can group strains based on their genetic profiles during outbreaks, which is particularly important in the context of identifying emergent resistant or virulent strains [192, 193].
Finally, while not directly related to clinical care, NGS is also advancing and improving procedures for public health related surveillance. WGS is currently being used alongside traditional methods for tracking and sourcing foodborne illnesses, showing improved speed and rates of detection [194–196]. Furthermore, NGS has proven valuable in monitoring the most prevalent strains of influenza, which has important impacts on the development of annual vaccines [197, 198].
Pathogen identification and diagnosis
Numerous examples in the literature describe the use of NGS to identify bacterial, viral, or eukaryotic pathogens in a wide range of sample types including synovial fluid, CSF, feces, corneal tissue, blood, plasma, and nasopharyngeal swabs [199–206]. Rather than describing these routine applications of NGS in detail, we will instead highlight a few cases that show the advantages of using NGS tests as opposed to traditional laboratory practices.
Traditional diagnostic laboratory methods, such as culture-based or PCR tests, are generally reliable and cost-effective for common pathogens; however, NGS and mNGS can specifically provide tremendous value in cases, where there is no a priori knowledge of the pathogen. One extreme example is a case of an adolescent with severe combined immunodeficiency who presented with infection symptoms for months without a diagnosis [207]. Nearly 40 different tests using multiple sample types, ranging from culture to PCR-based methods, proved non-diagnostic. Finally, the use of mNGS with CSF identified a very low number of reads that matched to Leptospira santarosai, demonstrating proof of a specific bacterial infection. Following this discovery, the patient was administered the appropriate antibiotics and responded to the treatment.
In another case, an mNGS approach was used to identify a likely cause of death in three men, who developed nearly identical CNS symptoms before dying, but showed no evidence of an infectious pathogen using multiple diagnostic tests [208]. The three men were squirrel breeders, had shared squirrels for breeding, and all three had previously been exposed to bites or scratches. Through mNGS of multiple tissue types isolated from a squirrel handled by one of the patients, the researchers identified a low level of sequences that corresponded to the bornavirus family, though this virus was ultimately determined to be novel based on phylogenetic studies. The bornavirus was confirmed using quantitative reverse transcription PCR in both the squirrel and patient brain tissue, as well as by immunohistochemical staining of patient tissues. There are multiple other examples of cases, where NGS found an unexpected or previously undiagnosed pathogen including eukaryotic organisms such as amoebae [209, 210].
In many settings, fast detection of the pathogen, as well as its associated resistance or virulence markers, is extremely important for appropriate and timely treatment; rGS can identify treatment options faster than conventional methods. Nanopore sequencing, which is as fast, or faster than standard approaches, has been used to identify pathogens, as well as antibiotic resistance markers [211, 212]. At one institution, Nanopore sequencing was found to shorten the average time to appropriate antibiotic therapy in pneumonia patients by roughly 24 hrs compared with standard methods, while delivering results in eight hrs post sequencing [213]. NGS is particularly helpful in situations where results are delayed or are non-diagnostic by culture, and can detect antimicrobial resistance or virulence markers at low frequencies, while still demonstrating sensitivity and specificity comparable to standard practices [172, 214–218].
NGS can also resolve discrepancies between standard culture-based and molecular-based diagnostic approaches and identify multiple organism co-infections, which may confound standard testing results [219, 220]. Furthermore, culture may be less effective as a diagnostic test when used to identify pathogens from patients already treated with antibiotics; in one study NGS showed significantly higher sensitivity than culture methods in patients with prior antibiotic exposure [221]. Finally, NGS using cell-free DNA in urine or blood has proven effective for diagnosing additional pathogens in cases where culture-based methods have failed [175, 204, 205].
Integrated sequencing of pathogens and the host
Up until now, we have focused on the sequencing and identification of infectious organisms. However, using datasets of the host’s own microbiome, as well as changes in host gene expression, can greatly aid the predictive value of testing. One study examining lower respiratory tract infections showed that an approach combining the gene expression signature of a patient’s immune response measured by profiling the host transcriptome via RNA-sequencing, alongside mNGS to identify and discern between the patient’s own commensal flora and pathogen genomes, was able to accurately identify the causative pathogen and achieve high sensitivity and specificity, with a true negative predictive value of 100% [222]. In addition to aiding in the interpretation of diagnoses, the host microbiome can also provide insight into the general wellness of a patient. For example, virome sequencing in immunocompromised patients post-organ or stem cell transplant can gauge the competency of the host immune system, as viral load can increase with use of immunosuppressant drugs [223–225]. Changes in the diversity of commensal bacterial flora can highlight disease onset or progression [226, 227]. Conversely, rescue of that diversity has been monitored using NGS by comparing results from a phylogenetic microarray alongside improved symptoms in patients with Clostridium difficile infections who were administered treatments such as fecal transplants [228]. Overall, the application of NGS has demonstrated diagnostic utility in infectious diseases, and further advancements in sequencing technologies will open new avenues for integrating both host and pathogen sequencing data, to provide a more holistic diagnosis and inform clinical management.
Go to :
CHALLENGES AND FUTURE PERSPECTIVES
Commonly used NGS technologies have limitations such as short reads and relying on clonal PCR to generate enough signals for detection. New technologies with long reads and single molecular sequencing (e.g., Pacific Biosystems and Oxford Nanopore) would theoretically be better and require less starting material; however, their high error rate prevents them from becoming the method of choice. GS is predicated to play an increasingly important role in laboratory medicine, as it does not require an upfront enrichment step and produces uniform coverage of the whole genome; however, data analysis, especially structural variant analysis, variant interpretation, and data storage, remains arduous in GS [229, 230]. In oncology, cancer heterogeneity is a challenge for sampling, variant detection, variant interpretation, and treatment recommendations. New methods, such as single cell sequencing [231] and liquid biopsy, are promising for addressing this issue. Germline alterations are also confounding issues in somatic cancer diagnostics; sequencing matched cancer and normal tissues simultaneously from the same patient is a plausible solution, although obtaining matched normal tissue has proven difficult in certain clinical situations [232].
The implementation of NGS tests in clinical diagnostic laboratories requires many resources [233]. Test validation, bioinformatics support, and data storage according to the guidelines are required before NGS test implementation and these are cost prohibitive for many small laboratories. Additionally, the current cost of clinical NGS tests remains high, limiting the usage of large panel testing, ES, and GS in cancer. Another obstacle of NGS application in laboratory medicine is insurance coverage. In 2018, the CMS finalized the National Coverage Determination, which covers NGS tests for patients with advanced cancer (https://www.cms.gov/medicare-coverage-database/details/nca-tracking-sheet.aspx?NCAId=296). In 2019, the CMS proposed a new national coverage policy for germline NGS panels for cancer patients, which will be finalized in 2020 (https://www.cms.gov/newsroom/press-releases/cms-expands-coverage-next-generation-sequencing-diagnostic-tool-patients-breast-and-ovarian-cancer).
Go to :
CONCLUSIONS
NGS is a breakthrough technology that opens new opportunities for molecular diagnostics. Many clinical laboratories have already adopted NGS technology to identify causal variants for the diagnosis of constitutional disorders, genomic profiling for precision oncology, and pathogen detection for infectious diseases. The NGS technologies and bioinformatics tools will continue to evolve and become the major diagnostic means and standard of care for genomic analysis to meet the ever-increasing demands of precision medicine.
Go to :
 XML Download
XML Download