

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Wilson disease (WD; OMIM #277900) is an autosomal recessive disorder associated with ATP7B variants. WD can manifest as multi-organ disturbances due to excessive copper accumulation and symptoms usually present between the ages of five and 35 years. Early manifestations of WD are easily overlooked and misdiagnosed as hepatitis, cirrhosis, splenomegaly, or encephalitis, leading to delayed treatment. If patients are diagnosed early and managed properly, permanent organ damage can be effectively prevented [1–3]. Asymptomatic or atypical WD diagnosis is still difficult, and few reports have diagnosed WD cases using next generation sequencing (NGS)-based gene panel test [4, 5]. We report a case of atypical WD mimicking spinocerebellar ataxia (SCA), diagnosed using an NGS panel test. Written informed consent for genetic test was obtained from the patient, his parents, and sister. This study was approved for waiver of informed consent by the Institutional Review Board of Kangbuk Samsung Hospital, Seoul, Korea (KBSMC 2018-10-018).
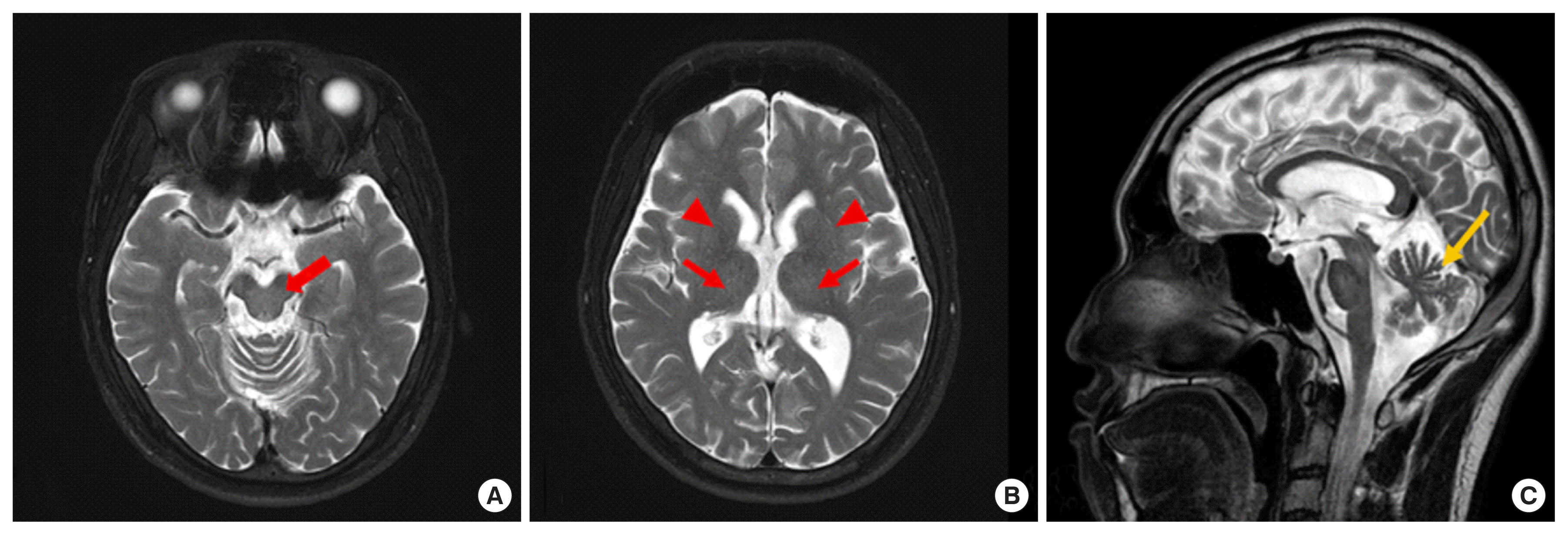
A 30-year-old male presented with a six-month history of bradykinesia and progressive gait instability. The neurological examination at admission on May 2018 indicated that he had limb ataxia, cerebellar type dysarthria, gait disturbance, and gait instability. Serum levels of aspartate aminotransferase (AST, 0.55 μkat/L), alanine aminotransferase (ALT, 0.48 μkat/L), total bilirubin (t-bil, 12.5 μmol/L), and gamma-glutamyltransferase (GGT, 1 μkat/L) were all normal. Brain magnetic resonance imaging (MRI) showed mild cerebellar atrophy without typical MRI findings, such as hyperintense lesions involving the putamen, pons, midbrain, and the thalamus, on the T2-weighted image (Fig. 1) [6]. Based on the cerebellar atrophy, MRI finding, and a history of bradykinesia and progressive gait instability, SCA was suspected.
Genetic testing of the SCA type 1, 2, 3, 6, 7, and 8 gene profile and DRPLA, as well as fluoro-propyl-carbomethoxy-iodophenyl-tropane positron emission tomography were conducted; all results were normal. In addition, an ataxia panel testing was conducted to distinguish other rare genetic diseases and hereditary ataxia.
Genomic DNA was extracted from the patient’s peripheral blood. The targeted gene panel included 293 genes spanning a 1,159,873 base pair (bp) region based on the hg19 human reference genome (https://www.ncbi.nlm.nih.gov/assembly/GCF_000001405.13/). Exomes were captured using a customized Target Enrichment System Kit (Celemics, Seoul, Korea). NGS was performed using the MiSeqDX platform (Illumina Inc., San Diego, CA, USA) with 150 bp paired-end sequencing, using the following pipeline: Burrows-Wheeler Aligner MEM algorithm 0.7.12 (https://arxiv.org/abs/1303.3997), Genome Analysis Toolkit 3.5 [7], Variant Effect Predictor (https://asia.ensembl.org/info/docs/tools/vep/index.html), dbNSFP (http://varianttools.sourceforge.net/Annotation/dbNSFP), SAMtools 0.1.19 (https://sourceforge.net/projects/samtools/files/samtools/0.1.19/), FreeBayes 0.9.2.1 (https://github.com/ekg/freebayes), and Scalpel 0.5.3 (http://scalpel.sourceforge.net/) and Picard tools 1.96 (https://broadinstitute.github.io/picard/) for data analysis. The NGS results identified a homozygous missense variant, NM_000053.3: c.3104G>T (p.Gly1035Val), in exon 14 of ATP7B, and this variant was classified as likely pathogenic.
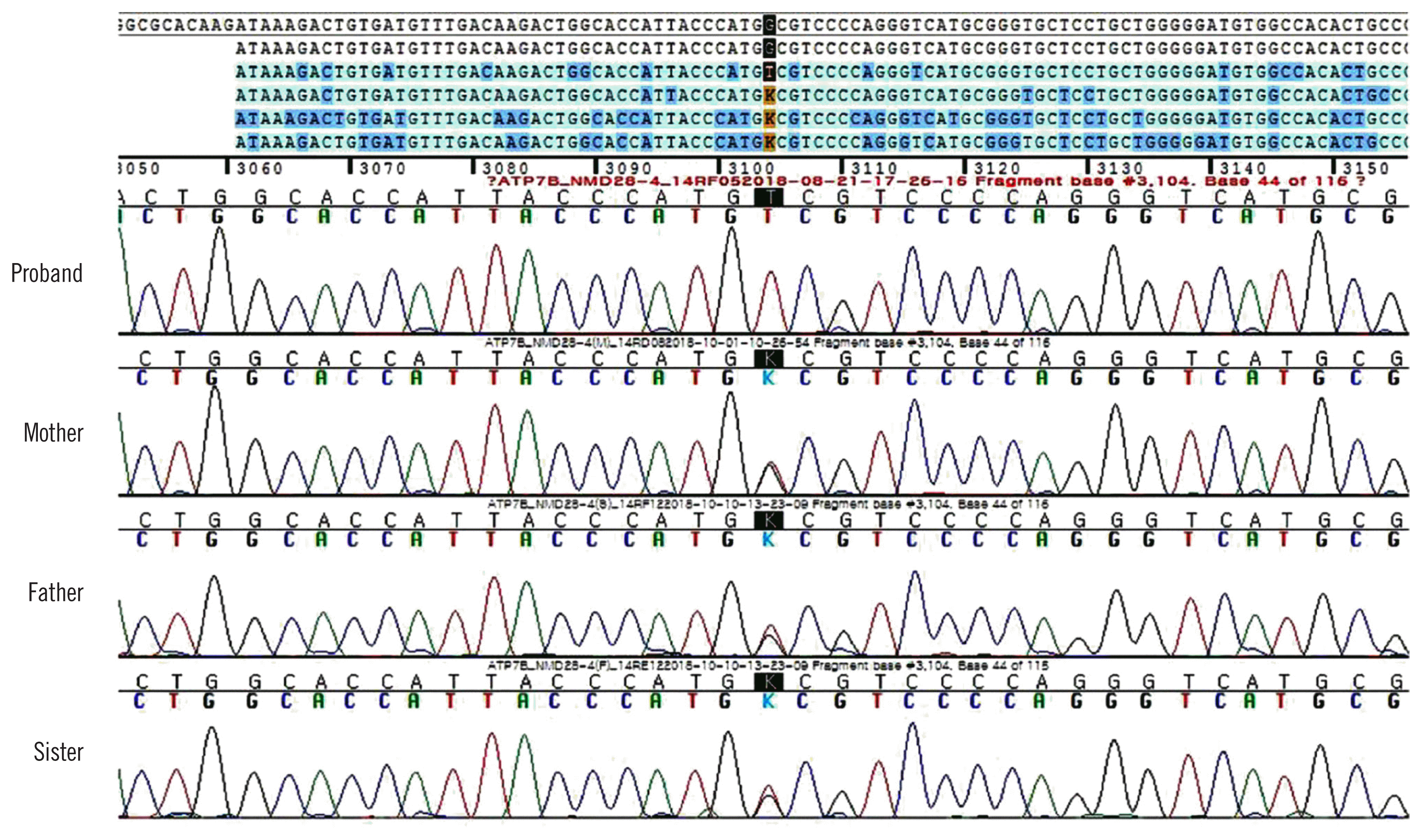
Although we did not suspect WD during the clinical work-up owing to late onset age, normal AST, ALT, and MRI findings, WD was genetically diagnosed based on the ataxia panel testing performed by NGS. After two months, the results of the additional tests we performed were as follows: the presence of Kayser-Fleischer (KF) rings under slit-lamp examination, decreased serum ceruloplasmin (45 mg/L [reference range, 200–600 mg/L]) and serum copper levels (4.4 μmol/L [10.7–22 μmol/L]), elevated 24-hr urine copper level (20.6 μmol/L [2.4–9.4 μmol/L]), and normal AST (0.52 μkat/L), ALT (0.5 μkat/L), t-bil (14.4 μmol/L), and r-GGT levels (1.06 μkat/L). Additionally, we re-evaluated the WD score following genetic test confirmation; the total WD score was 10 points (two points for the presence of KF rings, two for higher urine copper level, two for decreased serum ceruloplasmin level, zero for the absence of typical WD brain MRI result and hemolytic anemia, and four for the presence of the variant on both chromosomes) [8]. Finally, the patient was genetically and clinically diagnosed as having WD. Sanger sequencing of ATP7B among the patient’s family revealed that his father, mother, and sister harbored a heterozygous c.3104G>T variant and were asymptomatic carriers (Fig. 2).
The patient received oral penicillamine therapy at a dosage of 250 mg/day from September 2018, and his neurological symptom improved after treatment. The patient has been followed-up for one year under outpatient care with penicillamine therapy. One year later, the patient’s 24-hour urine copper level is within the stable range, and his health condition is good without any side effects.
WD is known to have a higher genetic prevalence than clinical prevalence, with a carrier frequency of approximately 1:90 based on the Hardy-Weinberg equilibrium; approximately 40% of WD patients have an obvious neurological presentation, ranging from movement disorders or tremors to rigid dystonia-like gait disturbance [9]. In Korea, the frequency of WD and its genetic carriers has been reported as 1:53, higher than in other countries [10]. In addition, the disease may be underdiagnosed, suggesting the need for WD screening in high-risk populations [10]. An NGS panel including ATP7B might be helpful for the differential diagnosis of neurological diseases.
NGS was a useful and reliable diagnostic method in our atypical WD patient. We suggest that NGS could be helpful in the case of atypical WD phenotypes. As WD can present with cerebellar ataxia and without Parkinsonism, brain MRI abnormalities, or elevated liver enzyme levels, it should be considered in all young patients with atypical hepatic or neurological symptoms. Moreover, it is important to include WD-associated genes when constructing a neurogenetic panel.
 XML Download
XML Download