

 PDF
PDF Citation
Citation Print
Print



Dear Editor,
Congenital chloride diarrhea (CCD; MIM#214700) is a rare, autosomal recessive disorder caused by pathogenic variants in the solute carrier family 26 member 3 (SLC26A3; MIM#126650) gene located on chromosome 7q31. SLC26A3 encodes an apical epithelial Cl-/HCO3- exchanger located in the ileum and colon [1]. The primary defect of the intestinal Cl-/HCO3- exchanger, which is coupled with a Na+/H+ exchanger, leads to persistent Clrich diarrhea from birth and life-threatening metabolic alkalosis [1]. Recently, a potential association between CCD and inflammatory bowel disease (IBD) has been proposed in Europe [2–4]. However, to date, there have been no reports of IBD development in East Asian patients with CCD. This is the first case of Crohn’s disease (CD) developed in a child of East Asian ancestry with genetically confirmed CCD. This study was approved by the Institutional Review Board of Samsung Medical Center (Number 2018-02-007) and was conducted at the Department of Pediatrics, Samsung Medical Center, Seoul, Korea, in 2018. Informed consent was obtained from the patient and his parents.
The patient is a Korean male who was born at 36+6 weeks of gestation in November 2007. Small bowel dilatation and maternal polyhydramnios were noted on fetal ultrasonography. The patient developed abdominal distension and watery diarrhea at birth, with a rate of 10–12 times per day. Laboratory tests revealed hypochloremic metabolic alkalosis and hypokalemia; the fecal Clwas elevated to 126 mmol/L. Direct sequencing of the SLC26A3 gene revealed two compound heterozygous variants: a previously reported variant at intron 12 (NM_000111.2:c.1407+3A >C) [5] and a novel missense variant (NM_000111.2:c.2135C>A;p.Thr712Lys), which have not been reported in CCD to date. Genetic testing of the patient’s parents revealed that both were heterozygous carriers for the variants (Fig. 1A).
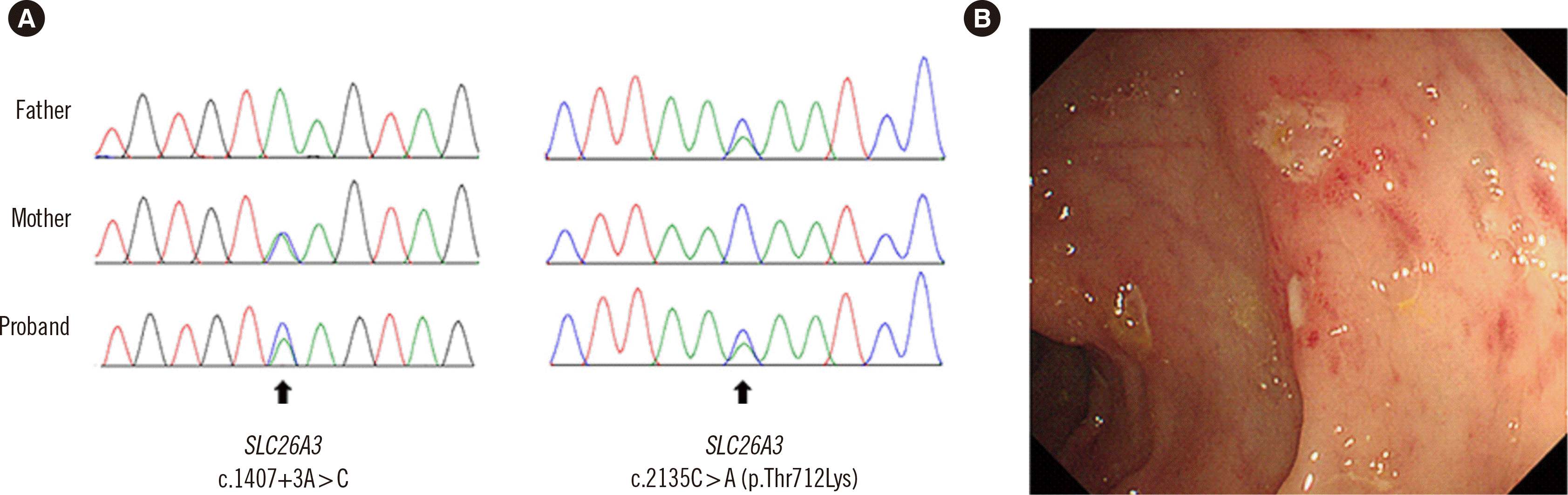
According to the Sequence Interpretation Guidelines of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology [6], the c.1407+3A > C variant could be classified as a likely pathogenic variant (LPV) based on the following evidence: it is absent in the gnomAD population database (PM2), listed as a deleterious mutation in the Human Gene Mutation Database (PP5), and predicted to disturb a consensus splice donor site based on three in silico splice site analysis algorithms (PP3; SpliceSiteFinder, MaxEntScan, and NNSplice) using Alamut Visual v.2.11 (Interactive Biosoftware, Rouen, France). In addition, the PM3 evidence code could be applied to this variant, as the patient reported by Hong, et al. [5] was a compound heterozygote for the founder pathogenic variant (c.2063-1G > T) and the c.1407+3A > C variant. Lastly, the PP4 evidence code could be applied based on the existence of two patients with CCD, including the present patient and the patient reported by Hong, et al. [5].
The c.2135C > A (p.Thr712Ly) variant could also be classified as an LPV based on the following evidence: it is absent in the gnomAD population database (PM2); predicted as a deleterious variant by in silico prediction tools (PP3) using PolyPhen-2 (probably damaging with a score of 1.000), SIFT (deleterious with a score of 0), and MutationTaster (disease causing with probability of 1); trans with the c.1407+3A > C variant (PM3); and has highly specific patient phenotypes (PP4) [6]. The patient’s hypochloremic metabolic alkalosis and hypokalemia were controlled by administration of salt substitution therapy with NaCl and KCl.
At the age of eight years, the patient developed chronic diarrhea, abdominal pain, and intermittent fever. Weight loss of 2 kg over two months was observed without any decrease in growth velocity. Laboratory tests showed a white blood cell count =8.44×109/L, hemoglobin =100 g/L, platelet count=465 ×109/L, erythrocyte sedimentation rate=77 mm/hr, C-reactive protein=41.2 mg/L, and albumin=39 g/L; the other laboratory tests were unremarkable. The fecal immunochemical test was positive, and the fecal calprotectin level was > 1,000 μg/g. No pathogens were detected in the stool culture and molecular testing. Ileocolonoscopy revealed multiple aphthous ulcers from the cecum to the rectum (Fig. 1B). The upper gastrointestinal endoscopy findings were unremarkable. Chronic inflammation with cryptitis was observed by histology. Magnetic resonance enterography revealed enhanced wall thickening in the distal descending colon, sigmoid colon, and rectum, without evidence of structuring or penetrating complications. Pelvic magnetic resonance imaging showed perianal inflammation at the 3 and 12 o’clock direction. The patient was diagnosed as having CD with an A1a, L2, B1p, G0 phenotype according to the Paris classification [7]. The patient’s pediatric CD activity index score was 30, and simple endoscopic score for CD was 18.
The mechanism underlying the association between CCD and IBD has yet to be elucidated. A previous study has shown that pathogenic variants of SLC26A3 are associated with a lack of a firmly adherent mucus layer and barrier impairment in mice [8]. In addition, intestinal SLC26A3 mRNA expression has been shown to be downregulated both in patients with ulcerative colitis and animal models [9]. Recently, it has been reported that tumor necrosis factor-α acts reciprocally with SLC26A3 and reduces its expression, leading to intestinal inflammation [10]. Further studies are required to reveal the underlying mechanism responsible for the development of CD in CCD patients.
Go to : 
 XML Download
XML Download