

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hereditary leiomyomatosis and renal cell cancer (HLRCC; MIM 150800) is a rare autosomal dominant cancer predisposition syndrome characterized by the variable development of cutaneous leiomyomas, early-onset uterine leiomyomas (fibroids), and type 2 papillary renal cell cancer (RCC), which is now classified separately as HLRCC-associated RCC [1, 2]. HLRCC is caused by germline loss of function variants in the FH gene, which encodes the Krebs cycle protein fumarate hydratase (FH) [3]; FH pathogenic variants have been identified in approximately 70–90% of individuals with suspected HLRCC [4-6]. Over 200 families with HLRCC have been reported worldwide [4-11]; however, the prevalence of HLRCC is unknown, and it may be underdiagnosed because of insufficient awareness of the condition and incomplete penetrance of HLRCC-associated tumors, especially RCC [6]. HLRCC-associated RCCs are found in only 20%–34% of affected individuals, while cutaneous and uterine leiomyomas occur in approximately 70–100% of patients with HLRCC by 30 years of age [12]. To date, no genotype–phenotype correlation has been identified for HLRCC [4, 6, 13].
In Korea, HLRCC and HLRCC-associated RCC have been only recently recognized, and data regarding the treatment outcome of HLRCC-associated RCC in a small series of genetically confirmed Korean HLRCC patients were reported in 2019 [10, 11]. We thus investigated the genetic spectrum of FH pathogenic variants in Korean patients with HLRCC, their phenotypic characterization, and the genotype–phenotype correlation. To the best of our knowledge, this is the first study to explore the clinical and genetic characteristics of Korean patients with HLRCC.
MATERIALS AND METHODS
Study patients
This study included Korean patients with suspected HLRCC at four academic hospitals in Korea (Gil Medical Center, Asan Medical Center, Seoul National University Hospital, and Pusan National University Yangsan Hospital) between December 2017 and August 2019. The selection criteria for the germline FH tests were as follows: histologically confirmed skin leiomyomatosis, suggestion of HLRCC-associated RCC based on clinicopathologic characteristics (young onset, non-clear cell RCC, including type 2 papillary RCC, a family history of RCC, very rapid progress, and unresponsiveness to standard treatments), early-onset symptomatic uterine leiomyomas in female patients, and a family history of HLRCC-associated symptoms. Five patients were recruited from a previous study by Choi, et al. [10] (Table 1). Demographic and clinical characteristics including age, sex, HLRCC-related phenotypes, and family history of participants were prospectively collected. Family members of genetically confirmed HLRCC patients were tested for germline variants of the FH gene regardless of symptoms.
Table 1
Clinical characteristics and FH variants of the 13 probands
| Case N | Sex | RCC (age, yr) | Hysterectomy or myomectomy (age, yr) | Family history | FH variant | |
|---|---|---|---|---|---|---|
|
|
||||||
| Identified variant | ACMG classification | |||||
| 1* | M | Yes (46) | NA | Uterine leiomyoma, cutaneous lesion | c.1108+1G > T | PV |
| 2 | F | Yes (40) | Yes (40) | Uterine leiomyoma | c.634C > T (p.Gln212*) | PV |
| 3* | M | Yes (42) | NA | No | c.132+1G > C | PV |
| 4* | M | Yes (29) | NA | RCC, uterine leiomyoma, cutaneous lesion | c.688A > G (p.Lys230Glu) | LPV |
| 5 | M | Yes (60) | NA | No | ND | |
| 6* | M | Yes (42) | NA | RCC, uterine leiomyoma | c.1027C > T (p.(Arg343*) | PV |
| 7 | M | Yes (34) | NA | No | ND | |
| 8 | F | Yes (64) | Yes (44) | Uterine leiomyoma | ND | |
| 9 | F | Yes (24) | No | No | ND | |
| 10* | M | Yes (49) | NA | No | c.842C > T (p.Thr281Ile) | VUS |
| 11 | F | Yes (30) | Yes (30) | Uterine leiomyoma | ND | |
| 12 | F | No | Yes (27) | RCC | c243dup (p.Gly82Trpfs*13) | PV |
| 13 | F | Yes (25) | Yes (25) | RCC, uterine leiomyoma | ND | |
*Patients included in the study by Choi, et al. [10]. Novel variants are shown in italics.
![]()
This study was approved by the Institutional Review Board of Gachon University Gil Medical Center, Incheon, Korea (GAIRB 2017-365). Written informed consent for the study was obtained from all participants.
Molecular genetic testing
Fresh peripheral blood samples were obtained and transported, under refrigerated conditions if necessary, to the molecular diagnostic laboratory in Gil Medical Center within 24 hours. We performed direct sequencing of the FH gene, including all coding exons and flanking intronic regions, using genomic DNA extracted from peripheral blood leukocytes (QIAamp DSP DNA Mini Kit; Qiagen, Valencia, CA, USA). Polymerase chain reaction (PCR) was performed using in-house designed primers (Supplemental Data Table S1). Direct sequencing was performed on an Applied Biosystems 3500xL Genetic Analyzer (Applied Biosystems, Foster City, CA, USA) using a BigDye Terminator v3.1 Cycle Sequencing Kit (Applied Biosystems). Sequence variants were identified based on GenBank reference sequences NM_000143.3 and NP_000134.2 using Sequencher 4.9 (Gene Codes Corporation, Ann Arbor, MI, USA). The identified sequence variants were interpreted and classified according to the 2015 American College of Medical Genetics and Genomics standards and guidelines [14]. Chromosomal microarray (CMA) was performed for female patients with negative sequencing analysis results who had both type 2 papillary RCC and uterine leiomyoma with a probable family history, using the CytoScan Dx Assay test with a resolution of 400 kb (Affymetrix, Santa Clara, CA, USA).
RESULTS
Clinical characteristics of the patients
The study included 13 probands (seven males and six females) and 19 family members. The demographic and clinical characteristics of the probands are presented in Table 1. Except one, they (12/13, 92.3%) were diagnosed as having RCC (nine papillary type, one mixed papillary and clear cell type, one clear cell type, and one unclassified). One female proband (P12) underwent a hysterectomy at 27 years of age, and a family history of type 2 papillary RCC was documented for her (in her father). Eight of the 13 (61.5%) probands had a family history of HLRCC-associated symptoms. Four of the six female probands had both RCC and uterine leiomyomas, and three of these underwent a hysterectomy before the age of 40 years. In addition, three of the seven (42.9%) male probands had female family members with a history of early-onset uterine leiomyomas. None of the patients in our case series had all three representative HLRCC tumors.
FH genetic testing
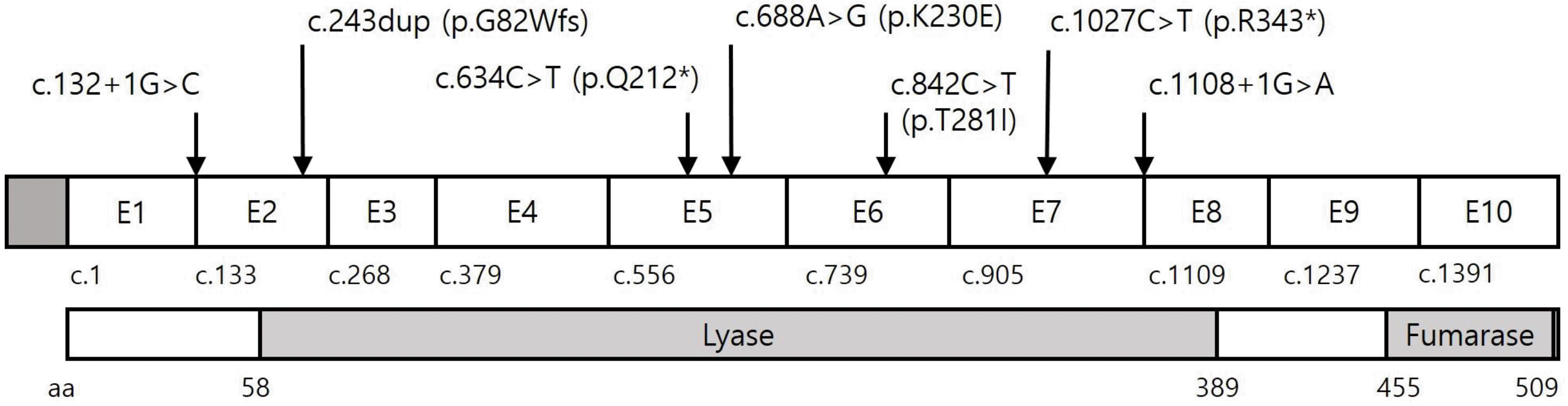
We found six different pathogenic or likely pathogenic FH variants in six of the 13 probands (46.2%) and one variant of uncertain clinical significance (VUS) in one proband (7.7%). The pathogenic or likely pathogenic variants included two nonsense variants, two splicing variants, one frameshift variant, and one missense variant; two (33.3%) of these variants were novel (Table 1). We found a novel missense variant in P10 [c.842C > T (p.T281I)], which was previously reported by Choi, et al. [10]. However, we classified the variant as VUS because it was also detected in three asymptomatic family members. All of these variants were located in the lyase domain (Fig. 1). Five genetically confirmed HLRCC probands (all except P3) had a family history of HLRCC-associated tumors. No significant variant was identified by direct sequencing analysis in six probands. Of these, three female probands (P8, P11, and P13) had both type 2 papillary RCC and uterine leiomyomas with a probable family history; however, no significant copy number variation (CNV) was detected by CMA testing. The other three probands had isolated type 2 papillary RCC without a definite family history.
We performed a targeted variant testing in 18 family members of the three probands with pathogenic FH variants (seven for P1, four for P3, and one for P12) and the proband with the VUS (six for P10) and found pathogenic variants in five of them. Of the seven family members of P1, three female carriers had undergone a hysterectomy before the age of 40 years (Fig. 2). The remaining two family members with a pathogenic FH variant (49-year-old brother from P1 and 24-year-old sister from P13) were asymptomatic at the time of genetic testing. Three of the six family members of P10 (with the VUS) had the same variant without any clinical symptoms. The detailed clinical and genetic characteristics of the family members who underwent targeted variant testing are described in Supplemental Data Table S2.
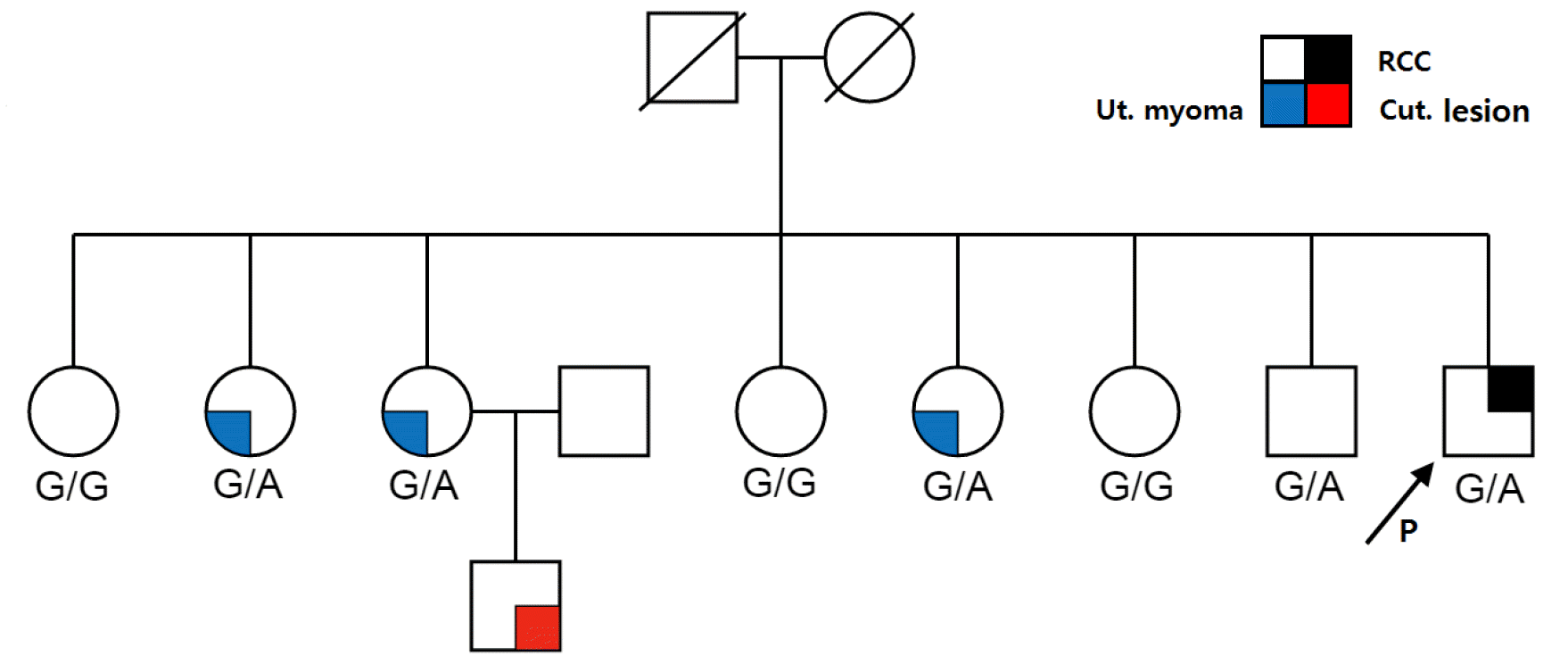
Fig. 2
Pedigree of P1 harboring the c.1108+1G > A variant (indicated by arrow). All three sisters of P1 who are carriers of the c.1108+1G > A variant underwent a hysterectomy before the age of 40 years. FH genetic testing was not performed for the son of the third sister. P1’s older brother was asymptomatic at the time of genetic testing.
Abbreviations: Ut, uterine; Cut, cutaneous.
![]()
Phenotypic characterization and genotype–phenotype correlation
All pathogenic and likely pathogenic variants were associated with RCC. Early-onset uterine leiomyomas were documented in all families with pathogenic or likely pathogenic FH variants, except in one proband (P3) whose mother and four sisters had neither HLRCC-associated symptoms nor a pathogenic FH variant. Cutaneous leiomyoma-like lesions were noted in the family members of P1 (c.1108+1G > A) and P4 [c.688A > G (p.K230E)], although a pathological diagnosis was not performed [11]. No significant genotype–phenotype correlation was observed.
DISCUSSION
We identified six families with HLRCC and six pathogenic FH variants, two of which were novel variants. Type 2 papillary RCC and early-onset uterine leiomyomas were frequently observed in the carriers of FH pathogenic variants, whereas cutaneous leiomyomas were relatively uncommon in our case series. In addition, we did not identify a definitive genotype–phenotype correlation.
In 2011, Smit, et al. [8] proposed criteria for the clinical diagnosis of HLRCC; according to these, diagnosis is likely when a proband has histopathologically confirmed multiple cutaneous piloleiomyomas (major criterion) or meets at least two of the following minor criteria: (1) surgical treatment for symptomatic uterine leiomyomas before the age of 40 years, (2) development of type 2 papillary RCC before the age of 40 years, and (3) has a first-degree family member who meets one of these criteria. Smit, et al. [8] observed 100% penetrance of cutaneous lesions in their study group aged > 40 years; however, none of the probands in our case series met the major criterion. All female carriers in our study underwent a hysterectomy before the age of 40 years, except one 24-year-old asymptomatic carrier, while only two probands were diagnosed as having type 2 papillary RCC before the age of 40 years (median 42 years; range, 29–46 years). Choi, et al. [10] reported that the median age of RCC diagnosis in 10 Korean HLRCC patients was 41 years (range, 27–52 years). Although Caucasian data have shown a high penetrance of cutaneous leiomyomas in HLRCC, such penetrance was less prevalent in Korean patients [5, 6, 8, 13]. Similarly, cutaneous manifestations were absent in Japanese and Chinese HLRCC patients [15-17]. It is likely that the phenotypic spectrum of HLRCC differs between Asian and Caucasian populations. There is a possibility of under-ascertainment of cutaneous leiomyomas as the participants of the current study mainly included patients with RCC who were recruited from the oncology department. It is also possible that our probands did not include patients with isolated uterine or cutaneous leiomyomas (formerly known as multiple cutaneous and uterine leiomyomatosis or Reed’s syndrome).
In the female carriers of FH pathogenic or likely pathogenic variants, uterine leiomyomas developed before the age of 40 years, with the earliest onset at 27 years of age. Early-onset uterine leiomyomas were the only manifestation noted in four of the seven female carriers at the time of the genetic testing. Uterine leiomyomas are the most common benign neoplasm in the female reproductive system; therefore, the condition itself is not likely to be taken into account when diagnosing HLRCC. To identify women with uterine leiomyomas who are at increased risk of HLRCC, pathology-based screening of uterine leiomyomas has been suggested, including FH-deficiency (FH-d) morphology (smooth muscle tumor cells with macronucleoli surrounded by a halo and cytoplasmic eosinophilic globules) and FH immunohistochemical staining [18-20]. Rabban, et al. [20] reported that 60% of women with an FH-d morphology had germline FH variants, and 0.24% of all women with any type of uterine leiomyoma were diagnosed as having HLRCC. In Korea, HLRCC has only recently been recognized by medical oncologists, while gynecologists and pathologists are unfamiliar with HLRCC-associated uterine leiomyomas. Therefore, our data demonstrate the necessity of pathology-based identification of candidates for HLRCC genetic testing among Korean women with early-onset uterine leiomyomas, which would enable clinical and radiological surveillance of HLRCC-associated RCC.
HLRCC-associated RCC has distinctive papillary architecture characteristics; however, none of the patients in our study were initially diagnosed as having HLRCC-associated RCC by a pathologist, as it is unknown to pathologists in Korea. In addition to a papillary pattern, a broad spectrum of architectural patterns has been reported for HLRCC-associated RCC, exclusive of clear cell type [7, 21, 22]. Therefore, ancillary tests, such as FH and 2-succinocystein immunohistochemical staining or a somatic/ germline variant test, will be helpful [7]. In a study of germline testing in patients with advanced RCC unselected for inherited syndrome risk factors, FH variants were the most common RCC-associated variants (seven of 41) [23].
Currently, approximately 100 pathogenic or likely pathogenic FH variants are listed in the ClinVar database (accessed December 2019). The variants include 78 null variants and 21 missense variants. Unlike the Caucasian data, which indicated that more than a half of the variants were missense variants, five of the six (83.3%) pathogenic/likely pathogenic variants in our study were null variants [4, 7, 13]. Large deletions, including that of the entire FH gene, have been identified in previous studies [3, 13, 24, 25]; however, we did not find any significant CNV in the three female patients who were highly suspected as having HLRCC based on the CMA test. Nevertheless, the possible presence of small exonal deletions cannot be ruled out as the detection limit of the CMA test used in this study was 400 kb. Additional high-resolution molecular tests, such as real-time PCR, multiplex ligation-dependent probe amplification, and high-definition arraycomparative genomic hybridization, are needed to detect small CNVs. It is possible that the FH sequencing-negative patients have other inherited cancer syndromes predisposing them to RCC, such as von Hippel-Lindau syndrome, hereditary papillary RCC, Birt-Hogg-Dubé syndrome, and tuberous sclerosis, although RCCs associated with these syndromes display distinctive clinicopathological characteristics [26, 27]. RCC predisposition gene panel tests may help identify patients with germline variants of cancer-susceptibility genes for whom tailored therapies and cancer screenings are indicated.
Our detection rate of FH pathogenic/likely pathogenic variants was relatively lower than those observed in previous studies (46.2%; 6/13; Table 2). The selection criteria we used for the germline FH tests were less strict than those used by Smit, et al. [8], indicating that patients less likely to have HLRCC were enrolled. Contrarily, our data showed that the HLRCC diagnosis criteria proposed by Smit, et al. [8] are less relevant to Korean patients, as none of the patients in our study met the major criterion, histopathologically confirmed multiple cutaneous piloleiomyomas. Thus, we suggest that FH genetic testing should be considered in Korean patients (1) who have non-clear cell RCC (especially with a type 2 papillary pattern) with a family history of RCC or early-onset uterine leiomyomas and (2) female patients with early-onset ( < 40 years old) uterine leiomyomas involving multiple family members, regardless of the presence of RCC and/or cutaneous leiomyomas.
Table 2
Frequency of FH PV/LPVs in various populations
| Reference | Geographic distribution/Ethnic background | Frequency of FH PV/LPV, % (N) |
|---|---|---|
| Toro, et al. (2006) [5] | North America | 93 (52/56 families) |
| Gardie, et al. (2011) [6] | France | 55.7 (44/79 families)* |
| Alam, et al. (2003) [7] | United Kingdom | 76.1 (35/46) |
| Smit, et al. (2011) [8] | The Netherlands | 42.4 (14/33 families) |
| Bhola, et al. (2018) [9] | Canada | 69.5 (48/69) |
| Present study | Korea | 46.2 (6/13) |
![]()
This study has potential limitations; it included a small number of patients from only four academic hospitals. Further, no central pathology review was conducted. Finally, sequencing-negative cases were not scrutinized using alternative molecular methodologies.
In summary, we describe the genotypic and phenotypic spectrum in a small series of Korean patients with HLRCC. Our data reveal the unique characteristics of Korean HLRCC patients, and the very low cutaneous leiomyoma penetrance. In addition, our data highlight the need for a more comprehensive approach for diagnosing patients with RCC and uterine/cutaneous leiomyomas who may benefit from tailored treatment and appropriate surveillance for cancer. Further studies on other Korean patients with HLRCC are required to delineate the clinical characteristics and establish a diagnostic strategy.
 XML Download
XML Download