

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR
Neurofibromatosis type 1 was first described by Frederich von Recklinghausen in 1882. Neurofibromatosis type 1 is an autosomal dominant disorder characterized by brown skin macules, iris hamartomas (Lisch nodules), and skeletal deformities [1]. Neurofibromatosis type 1 is a relatively common inherited disorder that affects about one in 2,500 to one in 3,000 people worldwide, irrespective of sex or ethnic origin [2]. The neurofibromatosis gene, located on chromosome 17q11.2, encodes a 220 kDa cytoplasmic protein called neurofibromin. This protein functions, in part, as a negative regulator of the Ras proto-oncogene, which is a key signaling molecule in the control of cell growth. Individuals with neurofibromatosis type 1 are prone to developing benign and malignant tumors of the central nervous system and peripheral nervous system. Furthermore, other cancers such as glioma of the optic pathway, glioblastoma, malignant peripheral nerve sheath tumor, gastrointestinal stromal tumor, breast cancer, and acute myeloid leukemia are associated with neurofibromatosis type 1 [3]. The association between multiple myeloma (MM) and neurofibromatosis type 1 is rare.
We describe a case of IgG-k MM with neurofibromatosis type 1. A comprehensive review of the literature looking for clinical-biologic correlations helping in identifying some aspect of the pathogenesis of the disease improve the manuscript.
A 69-year-old female with anemia, monoclonal component, and back pain was found to have MM. The neurofibromatosis type 1 had been diagnosed 40 years before. Molecular testing for neurofibromatosis gene mutations was performed and the results showed that patient one was heterozygous for the c.6855C>A (Tyr2285Ter) mutation.
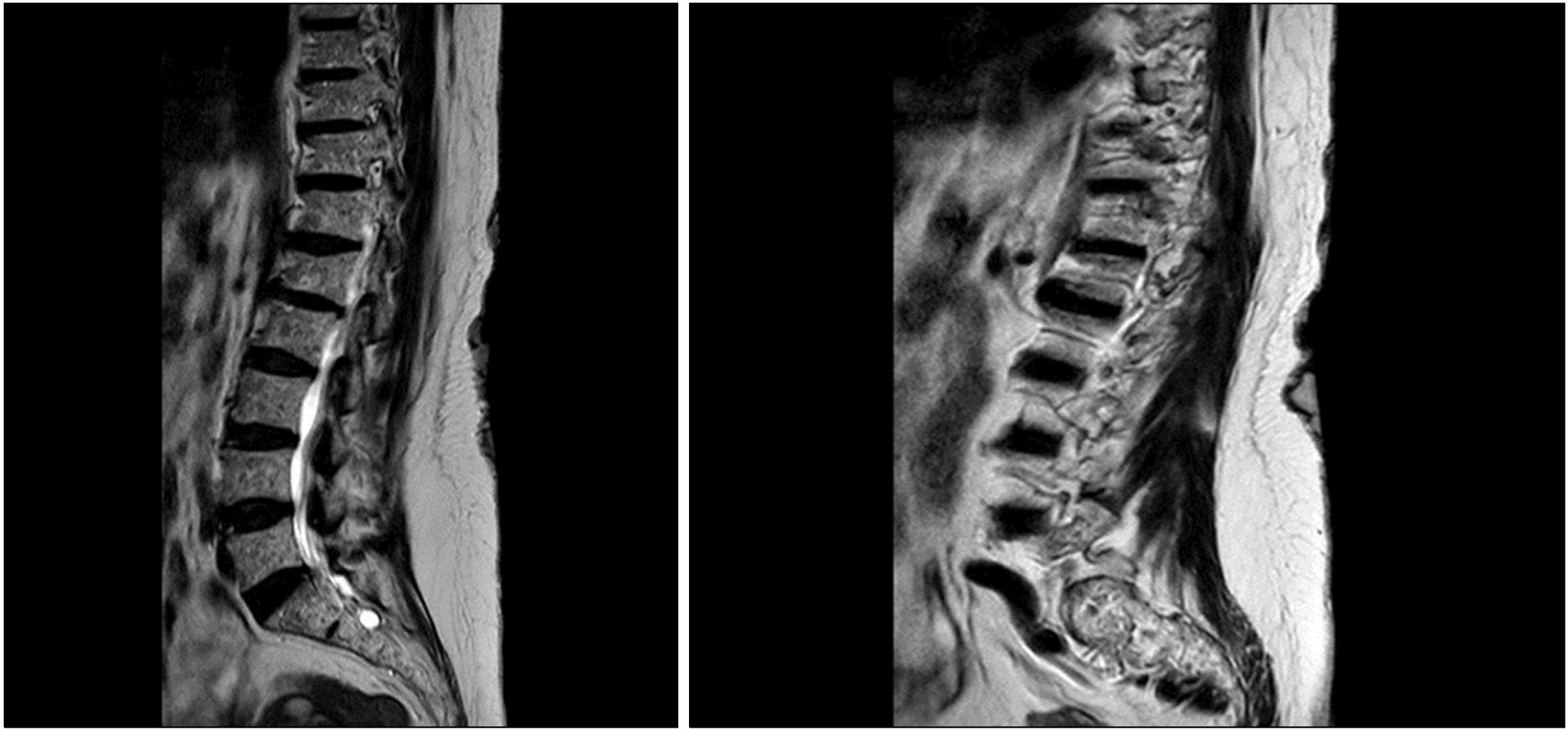
The mutation was not found in MMM cells. Serum immunofixation electrophoresis and Bence-Jones immunofixation on urine evidenced the presence of an IgG monoclonal component and kappa light chains, respectively. Protein electrophoresis showed a monoclonal spike with an IgG-k monoclonal component of 0.18 mg/dL. The determination of serum free light chains detected a kappa/lambda ratio of 168. The calcium and hemoglobin levels were normal. Fluorescence in situ hybridization (FISH) analysis of purified CD138+ plasma cells identified the presence of del(13q) but did not show del(17p). In addition, a bone marrow biopsy confirmed the presence of 70% plasma cell infiltration kappa restricted. A low dose computed tomography scan showed multiple collapses localized at dorsal and lumbar vertebrae. The magnetic resonance imaging revealed a collapse localized in the lumbar region based on the involvement of MM (Fig. 1).
Therefore, the patient was diagnosed with symptomatic IgG-k MM stage III according to the International Staging System (ISS) and was judged eligible for autologous stem cell transplantation. The first line of therapy consisted of six 28-day cycles of bortezomib, thalidomide, and dexamethasone (VTD regimen) [4], from June 2019 to November 2019. A stringent complete response was achieved. Subsequently, the patient underwent mobilization by cyclophosphamide plus granulocyte colony-stimulating factor followed by collection of peripheral blood progenitor cells; the patient is currently still waiting for the transplant procedure.
As far as we know, this is the fourth case of a possible association between MM and neurofibromatosis type 1 reported in the literature [5, 6]. However, the reason for this association is unknown. In our case, we have not observed a bad prognosis in this association. In Table 1,
Table 1
Summary of case reports on NF1 and MM.
| Case No. | Age/sex | Diagnosis | NF type |
Interval from NF1 and MM diagnosis |
Cytogenetic | Therapy | Outcome | Ref. |
|---|---|---|---|---|---|---|---|---|
| 1 | 59/M | MM IgG-Kappa | 1 | ND | Del 13q, tris 9 and 15 | VTD+auto-sct | VGPR | Accardi et al. [6] |
| 2 | 76/M | SMM IgG-Kappa | 1 | ND | ND | Follow-up | Accardi et al. [6] | |
| 3 | 69/F | MM IgG-Kappa | 1 | 40 yr | Del 13q | VTD | sCR | Our case |
| 4 | 63/M | MM light chain disease, Lambda | 1 | ND | (1q+), 8q+, 13q+,i (17q), i (18q), and +M. | ND | ND | Zengin et al. [5] |
![]()
The neurofibromatosis gene produces a GTPase-activating protein, which functions as a tumor suppressor. Either a duplication of 17q, containing the neurofibromatosis type 1 locus, or a loss of 17p containing p53 may provide the evaluation of tumor. Further, a normal or mutated neurofibromatosis type 1 gene may affect the other genes at the same locus or those in nearby loci and may contribute to tumorigenesis [7]. The increased frequency of neoplasms in neurofibromatosis type 1 patients is partly explained by the genetic basis of the disorder that results from mutations of the neurofibromatosis type 1 locus at chromosome 17. The neurofibromatosis gene is a tumor suppressor gene that encodes for neurofibromin, a GTPase-activating protein, which downregulates Ras [8, 9]. The neurofibromatosis gene mutations are likely to produce a non-functional neurofibromin protein, which results in the deregulation of Ras pathways. Recent studies have demonstrated that mutations in N-and K-ras oncogenes may play a major role in the pathogenesis of MM, which are probably more significant in the disease than previously thought [10]. Both neurofibromatosis and Ras genes participate in the signal transduction through Ras proteins [8]. The product of the Ras genes is a guanosine phosphorylated binding protein (Ras-GDP) which can be converted to the active GTP-bound form, leading to the activation of various signaling pathways that may affect growth, differentiation, and apoptosis. Mutations in Ras oncogenes promote the active GTP-binding protein, thus continuously activating downstream effectors and giving a growth advantage to malignant cells [11]. Mangues et al. [12] have found a significant increase in the incidence of lymphomas in animals that was attributed to the synergistic effect of N-ras overexpression and neurofibromatosis gene inactivation. Based on the above literature, the association of neurofibromatosis type I with plasma cell dyscrasia could be interpreted by common pathogenetic mechanisms through Ras pathways.
In addition, the International Myeloma Working Group consensus updated the definition for high-risk MM based on cytogenetics. Several cytogenetic abnormalities such as t(4;14), del(17/17p), t(14;16), t(14;20), non hyperdiploidy, and gain (1q) that confer poor prognosis were identified [13]. Currently, FISH is the standard technique for analysis of cytogenetic abnormalities. FISH is useful to detect genomic aberrations in situ and to enumerate the percentage of cells harboring such abnormalities, but it does not detect single-nucleotide variants. For example, TP53 on chromosome 17p is detected in 7% of myelomas, yet it is mutated at a much higher frequency in myelomas based on exome sequencing [14]. The neurofibromatosis gene is located in region 17q11.2, and the MM patients with this type of p53 (17p13) cytogenetic abnormality have a shorter progression free survival and overall survival [15]. Zengin et al. [5] highlighted the pathogenetic role of cytogenetic mutations in their patient, but the patient showed a complex karyotype without an overt association with a p53 mutation. The authors concluded that the patient had the same mutation located in the 17th chromosome that can cause neurofibromatosis type I and MM. On the contrary, Accardi et al. [6] reported a case of a 59-year-old MM patient for whom FISH analysis of purified CD138+ plasma cells identified the presence of del(13q) and trisomy of chromosome 9 and 15 but did not show del(17p) and chromosome 14 rearrangements. In the other case reported by Accardi et al. [6], the authors describe an association of smoldering multiple myeloma and neurofibromatosis type 1.
In conclusion, we suggest that the association between MM and neurofibromatosis type 1 may be a mere coincidence. However, during the work-up of MM in patients with neurofibromatosis, it is important to consider the possibility of del(17) that indicated a poor prognosis. Further studies are needed to deeply explore the possible link between MM and neurofibromatosis type 1.
Go to : 
 XML Download
XML Download