

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR
B-cell prolymphocytic leukemia (B-PLL) is an extremely rare malignancy (<1% of B-cell leukemias) [1, 2]. Given a wide array of differential diagnoses, establishing the correct diagnosis is challenging [3]. Histomor-phological evaluation of the peripheral blood (PB) smear and diligent interpretation of the immunophenotype of lymphocytes by an expert pathologist are essential for the diagnosis of B-PLL. In this report, we describe the case of a patient with B-PLL, discuss the diagnostic possibilities, and briefly review the relevant literature.
A 65-year-old man presented to our hospital in August 2019 with the complaints of fever and left upper abdomen pain for 6 months. General examination showed pallor. Abdomen examination revealed massive splenomegaly (12 cm below left costal margin). CT scan of the abdomen showed splenomegaly (23 cm), and multiple enlarged lymph nodes in the retroperitoneal, peri-portal, peri-epigastric, and pelvic areas. Complete blood count results showed hemoglobin level to be 70 g/L and white cell count at 470×109/L (including 99% lymphocytes and platelet count of 127× 109/L). Peripheral blood (PB) smear revealed 90% prolymphocytes (Fig. 1). Immunophenotypically, the lymphocytes were CD45+, CD5+, CD23+ (heterogeneous), CD200dim+, CD19+, CD20+ (bright), surface CD22+ (bright), surface CD79b+ (bright), CD38+, surface kappa+ (bright), and surface IgDdim+. Lymphocytes were negative for CD10, FMC7, CD25, CD11c, CD123, CD103, surface lambda, Immunoglobulin G (IgG), and Immunoglobulin M (IgM) (Fig. 2). Morphological and immunophenotypic findings were consistent with the diagnosis of de-novo B-PLL. PB interphase fluorescent in-situ hybridization (FISH) failed to reveal deletion 11q, deletion 6q, deletion 17p, trisomy 12, deletion 13q, and t (11,14) (q13; q32). Bone marrow (BM) biopsy was hypercellular, and showed complete replacement by prolymphocytes. Immunohistochemistry for cyclin D1 on BM biopsy was negative. Cytogenetic analysis of BM aspirate revealed a normal karyotype. Iron profile, serum vitamin B12, and folate levels were normal. Viral markers and direct antiglobulin test were negative. Serum lactate dehydrogenase was elevated (784 U/L; normal, <250 U/L). The patient was treated with BR chemoimmunotherapy [Bendamustine (90 mg/m2 on days 1 and 2) and Rituximab (375 mg/m2 on day-1)] administered every 28 d. Patient achieved complete remission (CR) after 6 cycles of BR, and continues to be in CR till date.
 | Fig. 1Microphotograph of the bone marrow aspirate smear showing almost complete replacement by prolymphocytes (2–2.5 times the size of a mature lymphocyte with mild-moderate cytoplasm, open chromatin, and prominent nucleoli, Giemsa–Wright stain, ×100).
|
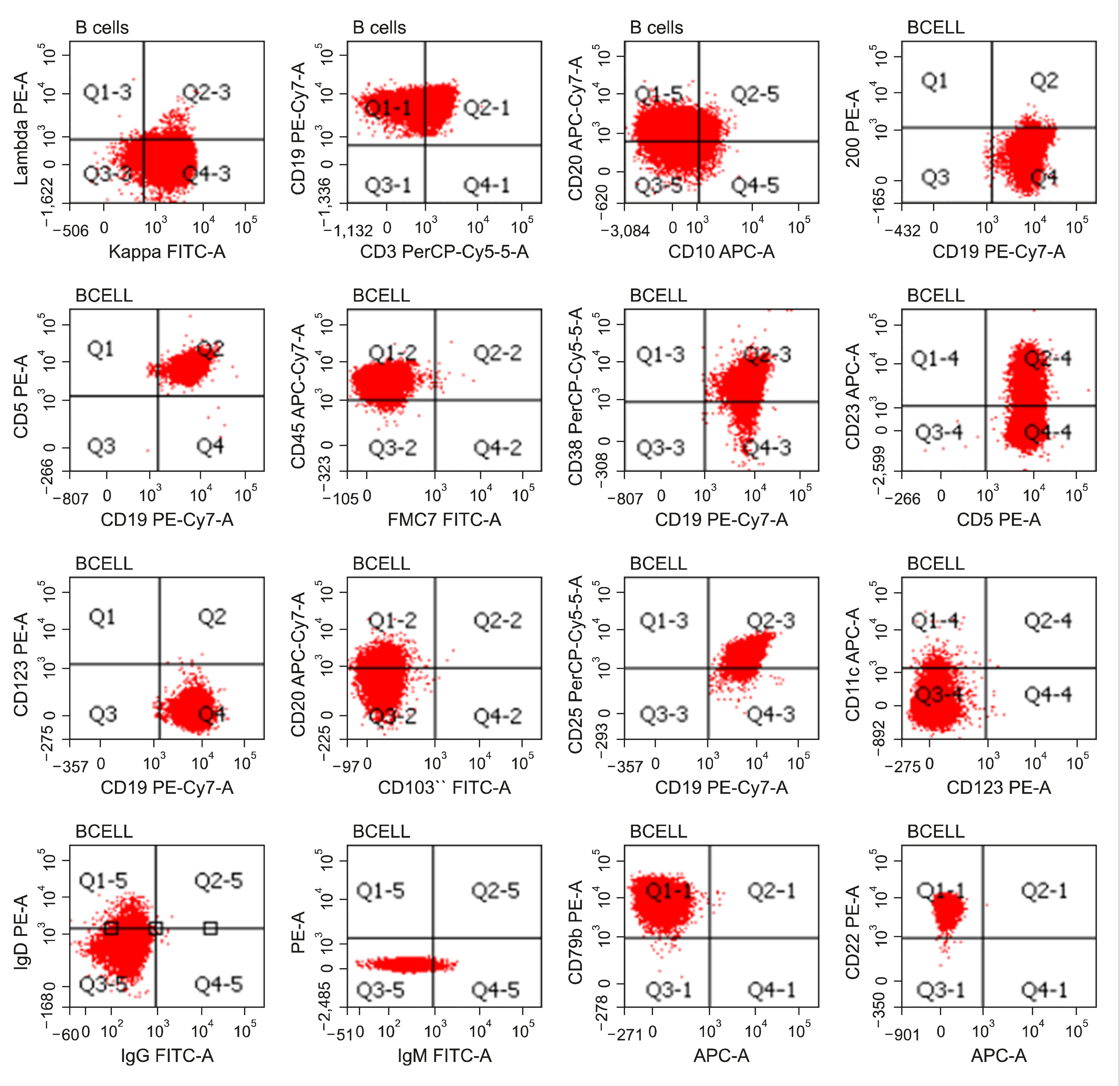 | Fig. 2Flow cytometry plots of the patient showing immunophenotype of the B-lymphocytes (red color). B-lymphocytes were CD45+, CD5+, CD23+ (heterogeneous), CD200dim+, CD19+, CD20+ (bright), surface CD22+ (bright), surface CD79b+ (bright), CD38+, surface kappa+ (bright), and surface IgDdim+. CD10, FMC7, CD25, CD11c, CD123, CD103, surface lambda, IgG, and IgM were negative.
|
PLL is defined as the presence of >55% prolymphocytes in the PB and BM. PLL has two subtype: T-PLL and B-PLL, the latter being much rarer [1-3]. Immunophenotypically, B-PLL shows bright expression of B-cell markers (CD19, CD20, FMC7) and surface immunoglobulins (sIg), negative expression of CD5, CD23, CD200, CD10, and T-cell markers, and demonstrates a low Matutes score [3]. Expression of CD5 and CD23 is uncommon (1/3rd cases) in B-PLL [1], and CD200 expression has occasionally been reported [4]. B-PLL could arise either de-novo or from prolymphocytic transformation of chronic lymphocytic leukemia (CLL). In cases of prolymphocytic transformation of CLL, prolymphocytes retain the immunophenotype of CLL, though they show a brighter sIg expression [1]. Old age (6th–7th decade), B-symptoms, massive splenomegaly, high white cell count, and anemia with/without thrombocytopenia are the classical clinical features of B-PLL. Peripheral lymphadenopathy is uncommon [2]. Cytogenetic abnormalities, c-myc aberration, and deletion 17p/TP53 mutation are seen in about 75%, 60%, and 40% cases, respectively [5]. Based on the c-myc aberration, and deletion 17/TP53 mutation, B-PLL is classified into three prognostic groups; low-risk (myc-activation- and deletion 17p-), intermediate-risk (myc-activation+ and deletion 17p-), and high-risk (myc-activation+ and deletion 17p+) [6]. Clinical course of B-PLL is aggressive. Prognosis is poor with median overall survival of about 2–3 years [2]. Due to its rarity and absence of prospective clinical trials, there are no formal guidelines for the management of B-PLL. Consensus regarding the treatment of B-PLL is mainly derived from anecdotal reports, and case series [7]. Patients with deletion 17p/TP53 mutations require targeted therapies like Bruton tyrosine kinase inhibitors (Ibrutinib), phosphoinositide 3-kinase inhibitors (idelalisib), and alemtuzumab (anti-CD52 monoclonal antibody). Patients without deletion 17p/TP53 mutations might benefit from chemo-immunotherapies (rituximab-based combinations with fludarabine or bendamustine). Allogeneic stem cell transplant should be considered either frontline in young and fit patients—particularly those with deletion 17p/TP53 mutations—or in relapsed/refractory cases [7-9]. Strong expression of B-cell markers, and surface kappa, heterogeneous CD23, and dim CD200 were against the diagnosis of CLL in our case. Presence of prolymphocytes, and negative expression of CD10 excluded follicular lymphoma. Lack of CD25/CD11c/CD123/CD103 ruled out hairy cell leukemia. Presence of classical prolymphocytes in our case did not support the diagnosis of marginal zone lymphoma, which usually shows the presence of villous lymphocytes on PB and BM. Negative interphase FISH for t (11,14) (q13; q32) and immunohistochemistry for cyclin D1 on BM biopsy helped exclude mantle cell lymphoma [10]. Our case had a negative expression of surface IgM and dim expression of surface IgD. Although B-PLL usually shows bright surface IgM+/-IgD expression, few cases of B-PLL similar to our case with typical prolymphocyte morphology, bright B-cell antigen expression, and without surface IgM/IgG expression have been reported [11-13]. We report the current case due to its rarity and potential diagnostic as well as therapeutic challenges. This report highlights that flow cytometry is suggestive; however, a combination of clinical, morphological, and immunophenotypical features is imperative to diagnose B-PLL.
Go to : 
 XML Download
XML Download