

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR
Cyanosis is an abnormal bluish discoloration of the skin and mucous membranes. It is most commonly seen in conditions associated with cardiovascular and respiratory insufficiency that lead to hypoxemia. A rare cause of lifelong cyanosis is methemoglobinemia occurring due to either an enzymatic deficiency or the presence of abnormal hemoglobin variants known as hemoglobin M (Hb-M) [1]. Herein, we report the case of a 2-year-old child who was found to have a rare Hb-M variant resulting from a de novo mutation.
The proband was a 2-year-old girl who presented at the pediatric hematology clinic with anemia and a bluish discoloration of the lips. Her parents indicated a history of fever and respiratory infection dating to three months ago and she was found to have pallor and cyanosis. All these findings were ascribed to the respiratory tract infection, and she was treated accordingly. The infection subsided; however, cyanosis continued for several weeks. The child had no history of fatigue or exertional dyspnea and no significant history of any recent drug intake. She was the first child born from the non-consanguineous marriage of the parents, had a birth weight of 3 kg, and had an uneventful neonatal period. The family history was unremarkable. Clinically, the child was active and playful but had pallor; her lips and nails showed bluish discoloration due to cyanosis (Fig. 1A). However, no clubbing or organomegaly was found. Her vital parameters were within normal limits. Echocardiogram revealed normal parameters. Other systems were also unremarkable.
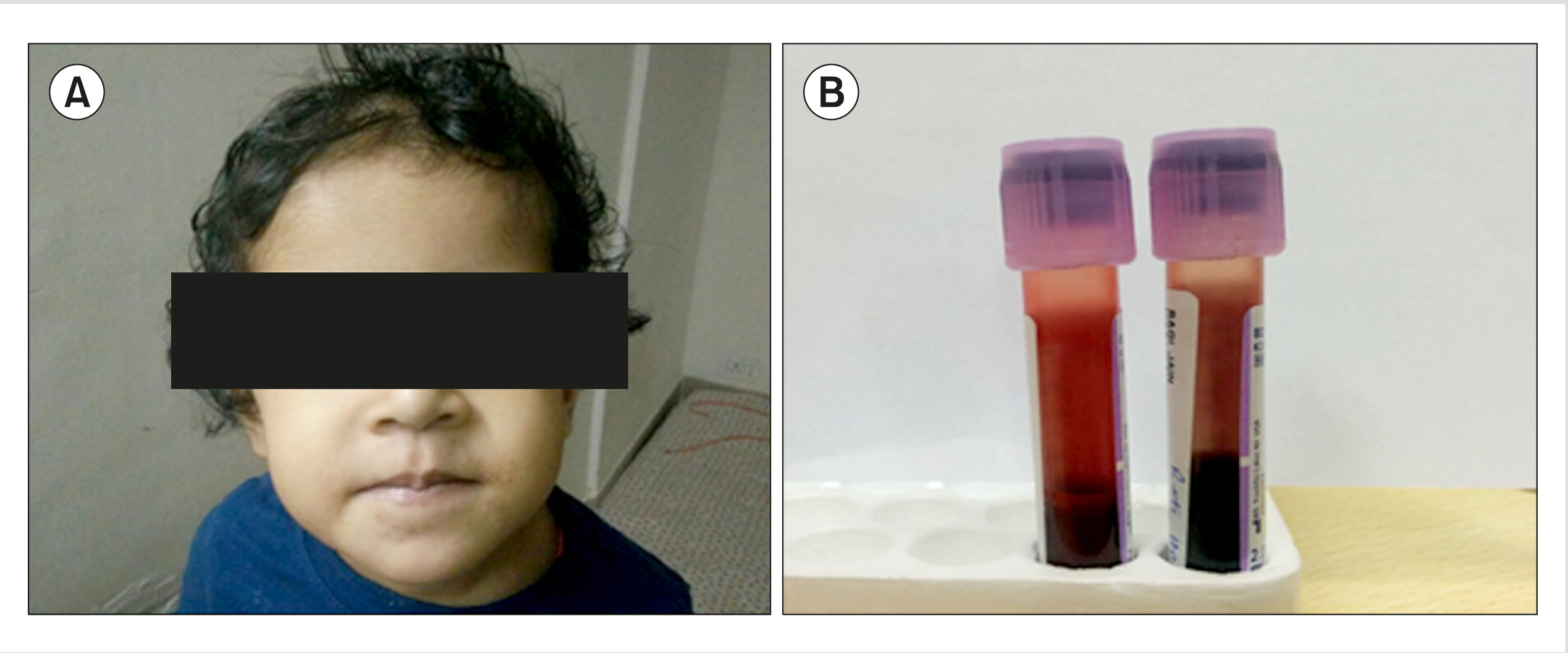
A peripheral blood sample was sent for routine hematological and biochemical investigation. Complete blood count showed the following findings: hemoglobin (Hb), 10.1 g/dL; red blood cell count, 4.14×106/μL; hematocrit, 32.0%; mean corpuscular volume, 77.3 fL; mean corpuscular hemoglobin (MCH), 24.4 pg; MCH concentration (MCHC), 31.6 g/dL; red cell distribution width, 16.9%; total leucocyte count, 13.2×103/μL; platelets, 496×103/μL; reticulocyte count, 1.1%. Biochemical tests such as serum iron profile, liver function test, and G6PD assay showed normal results. Oxygen saturation measured by pulse oximetry was 57%, while SaO2 was 98.1%. However, the methemoglobin level was high at 6.7% (normal, <1%). The blood sample also showed a distinct dark brown color (Fig. 1B).
To investigate the possibility of anemia, high-performance liquid chromatography (HPLC) was performed; the results revealed an unknown Hb peak of 14.3% in the C-window at a retention time (RT) of 5.06 min with increased Hb F levels (3.1%; normal, 0.1–1.2%) and elevated Hb A2 (4.0%; normal, 2.0–3.3%) (Fig. 2A). Considering the levels of Hb A2, Hb F, and abnormal hemoglobin, HPLC was carried out in samples collected from the parents, which showed normal levels. Thus, gene sequencing of the child and her parents was performed. Automated DNA sequence analysis of the beta globin gene in the patient showed a C>T substitution at codon 92 (Fig. 2B). This heterozygous point mutation leads to the substitution of histidine (CAC) by tyrosine (TAC) at the b 92 position, which is seen in Hb-M Hyde Park (also known as Hb Milwaukee 2). However, parent gene sequencing analysis showed normal results. Moreover, no other mutations for beta thalassemia trait were seen in both the child and her parents.
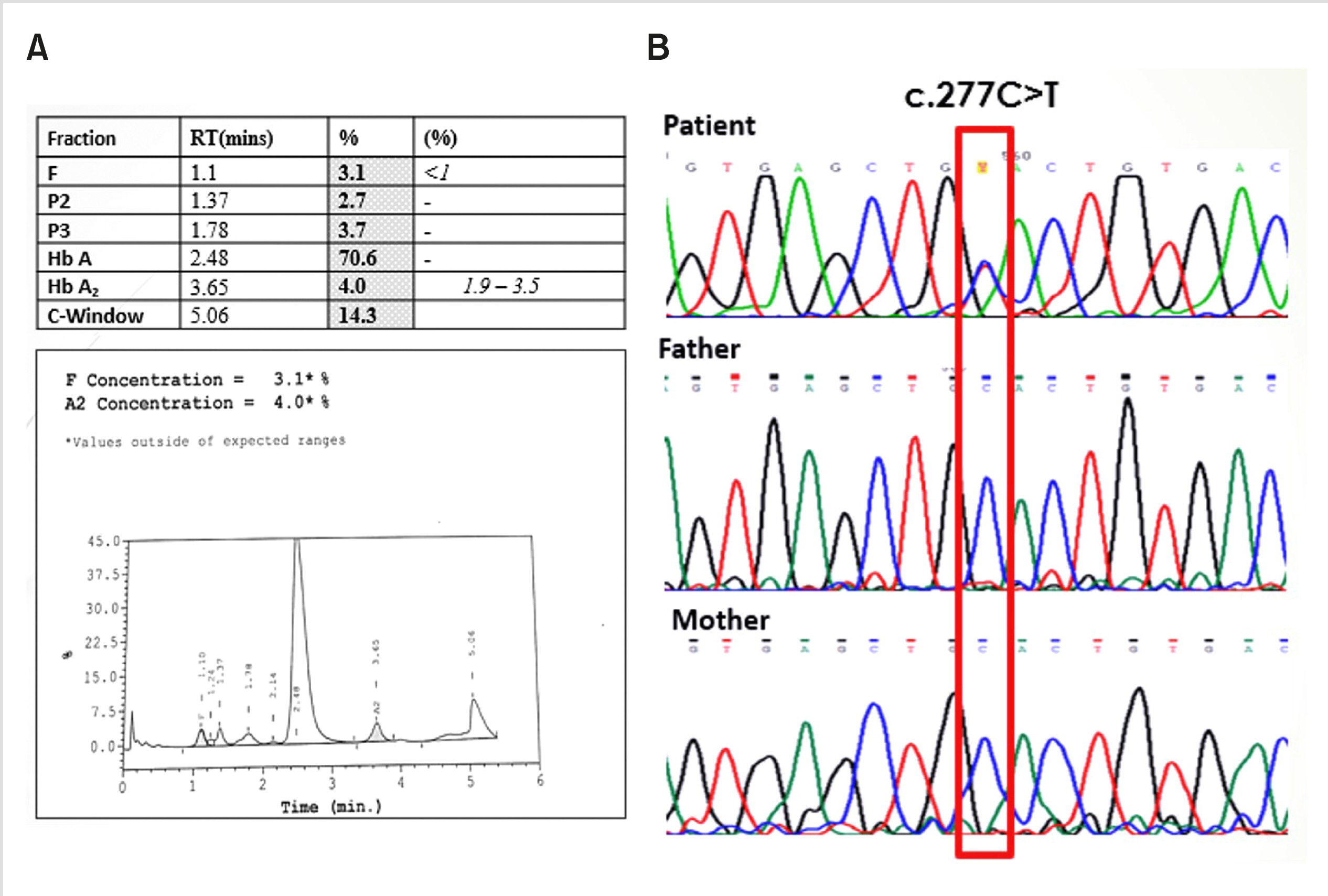
Considering the negative family history for cyanosis and the normal parental HPLC and DNA mutational analysis, the patient was diagnosed with Hb-M Hyde Park due to a de novo mutation.
Methemoglobinemia is a disorder in which there are increased levels of methemoglobin (MetHb) in the circulation. The iron in MetHb is present in the ferric state, which is much more stable than the ferrous state, and thus, it has a decreased ability to bind to O2 [1]. The most common cause of methemoglobinemia is the intake of certain drugs and chemical agents including local anesthetic agents such as lidocaine, procaine, and benzocaine; aniline dyes, nitrates, sulfonamides, primaquine, dapsone, and acetaminophen, among others. Some inherited conditions such as cytochrome B5 reductase deficiency and NADPH-methemoglobin reductase deficiency can also lead to this disorder [1, 2]. Methemoglobinemia may arise due to mutations in globin chains—alpha, beta, or gamma—leading to the formation of abnormal hemoglobin, Hb-M [1-3]. Hb-M displays an autosomal dominant inheritance pattern. It is a rare hemoglobinopathy in which the proximal or distal histidine in the alpha, beta, or gamma subunit is replaced by a tyrosine residue [1].
Hb-M Hyde Park/Hb-M Milwaukee2/Hb-M Akita is a rare beta globin chain variant of hemoglobin M wherein the mutation is present at codon 92 by C>T substitution leading to the replacement of histidine by tyrosine (His>Tyr) [1]. Patients with Hb-M Hyde Park are usually asymptomatic but present with cyanosis. This is an unstable hemoglobin resulting in a mild hemolytic picture with anemia and reticulocytosis. Owing to the prominent cyanosis, patients are frequently misdiagnosed for cyanotic heart disease [1].
The first case of Hb-M Hyde Park was reported in 1966 by Heller et al. [4] in black patient in the USA. Since then, very few cases have been reported worldwide. In 1968, Shibata et al. discovered a new variant of Hb-M in a family with hereditary cyanosis in the Akita province of Japan. It was named as Hb-M Akita but was later found to have the same primary hemoglobin structure as in Hb-M Hyde Park [5, 6]. Hutt et al. [7] in 1998 found Hb-M Milwaukee 2 in a patient with Hb E trait. They also found that the mutation in this case was identical to Hb-M Akita and Hb-M Hyde Park.
Most of these reported cases had a hereditary pattern in which the patient’s parents, siblings, or both had similar symptoms and were found to have Hb-M Hyde Park. However, only two cases of Hb-M Hyde Park arising from a de novo mutation have been described thus far. Stamato-yannopoulas et al. [8] in 1976 reported a case of a 10-year-old child who had cyanosis and Hb-M Hyde Park. However, none of the parents showed any symptoms or positivity for this Hb variant. Another case occurred in a 6-year-old girl who was diagnosed with Hb-M Hyde Park in 1992. Rotoli et al. [9] reported that neither the parents nor the sibling showed any abnormalities.
In India, only few cases of Hb-M have been reported, which includes Hb-M Iwate, Hb-M Ratnagiri, and Hb-M Boston [10-12]. However, only a single case of hereditary Hb-M Hyde Park has been reported in India to date. Upadhye et al. [12], in 2014, reported the case of a 26-year-old man who was incidentally found to have low oxygen saturation and cyanosis. On further investigation, he was found to have elevated methemoglobin levels. HPLC and DNA sequence analysis confirmed a diagnosis of Hb-M Hyde Park. Subsequently, his mother was also found to have similar complaints [12].
Therefore, to the best of our knowledge, this study is the first to report a case of Hb-M Hyde Park arising due to a de novo mutation in India. The patient in our case presented with the classical history of anemia with cyanosis: dark colored blood and elevated methemoglobin levels. HPLC and DNA sequencing results were indicative of Hb-M Hyde Park. Our patient showed elevated levels of HbF and HbA2, which was also observed by Loong et al. [13]. Although elevated levels of HbF and HbA2 are found in individuals with the beta thalassemia trait, our patient and her parents did not show any such mutation in their DNA sequencing. The cause of the elevated HbF and HbA2 in this patient remains unknown and needs further study. Hb-M Hyde Park patients are usually asymptomatic and thus require no further treatment. However, because Hb-M shows greatly decreased oxygen affinity compared to normal hemoglobin and this affects the oxygen saturation, its significance increases under general anesthesia [14].
To conclude, Hb-M Hyde Park is a rare hemoglobinopathy and should be considered as the underlying cause of cyanosis, especially when the search for other common causes has been unproductive. However, the importance of HPLC and DNA gene sequencing in the identification of abnormal hemoglobin variants is indispensable. Generally, the clinical course of such patients is unremarkable. However, owing to low oxygen affinity of hemoglobin M, these patients require special attention by an anesthetist when undergoing any surgery.
Go to : 
 XML Download
XML Download