

 PDF
PDF Citation
Citation Print
Print


TO THE EDITOR: Aplastic anemia (AA) is a hematological disorder that leads to significant mortality and morbidity. Clonal evolution in AA after immunosuppressive therapy (IST) is a major concern. Since Dameshek’s 1967 editorial in Blood, titled “Riddle: what do aplastic anemia, paroxysmal nocturnal hemoglobinuria (PNH) and ‘hypoplastic leukemia’ have in common?,” to genomic data have increased our understanding of clonal evolution; however, our knowledge remains incomplete [1, 2]. The earliest evidence came from reports of the clustering of myelodysplastic syndrome (MDS) and PNH in known cases of AA. Many case reports, series, and structured genetic studies have provided evidence of clonal hematopoiesis (>50%) and the development of clonal diseases in AA. Complications of clinically relevant PNH have been described in 15–25% of patients with accquired AA treated by IST. However, MDS and acute myeloid leukemia (AML) are reported in 5–15% of AA cases within 5–11 years [3]. The significantly increased risk of clonal evolution in the context of inherited bone marrow failure disorder (IBMF) is attributed to the nature of the genetic lesions of these syndromes [2].
Philadelphia positivity is a hallmark of chronic myeloid leukemia (CML) and well described in acute lymphocytic/lymphoblastic leukemia (ALL). BCR-ABL positivity in AML has been investigated and raised doubts regarding CML in blast crisis [4]. With emerging data, the World Health Organization (WHO) described Philadelphia (Ph)/BCR-ABL-positive AML in 2016 as a provisional entity under the heading “AML with recurrent cytogenetic abnormalities” [5]. Ph positivity in AML is a rare event and usually falls in one of the three types: AML-not otherwise specified, AML with myelodysplasia-related changes, and core-binding factor (CBF) AML, with Ph as an additional abnormality. A subset of Ph-positive AML has been reported during the evolution in known cases of MDS [6]. However, to the best of our knowledge, the development of Philadelphia-positive AML in AA has not been reported.
We hereby report this unusual case of clonal evolution to Philadelphia-positive AML in a case of AA on the wheels of PNH.
Case
A 17-year-old woman presented to our outpatient department in 2010 with chief complaints of fatigue and intermittent fever for 1.5 months. She also had a 1-week history of excessive bleeding per vaginum. Examination revealed petechiae on her hands and legs. Organomegaly, lymphadenopathy, and stigmata of IBMF were not observed. Complete blood count (CBC) analysis revealed pancytopenia (hemoglobin, 9 g/dL; total leukocyte count, 2.5×109/L; absolute neutrophil count, 0.4×109/L; platelet count, 5×109/L). Peripheral blood smear showed predominantly mature-appearing lymphocytes with normocytic normochromic RBCs. No blasts or abnormal cells were noted. Her corrected reticulocyte count was 0.2%. Serologic testing for hepatitis B surface antigen (HBsAg) and hepatitis C virus (HCV) was negative.
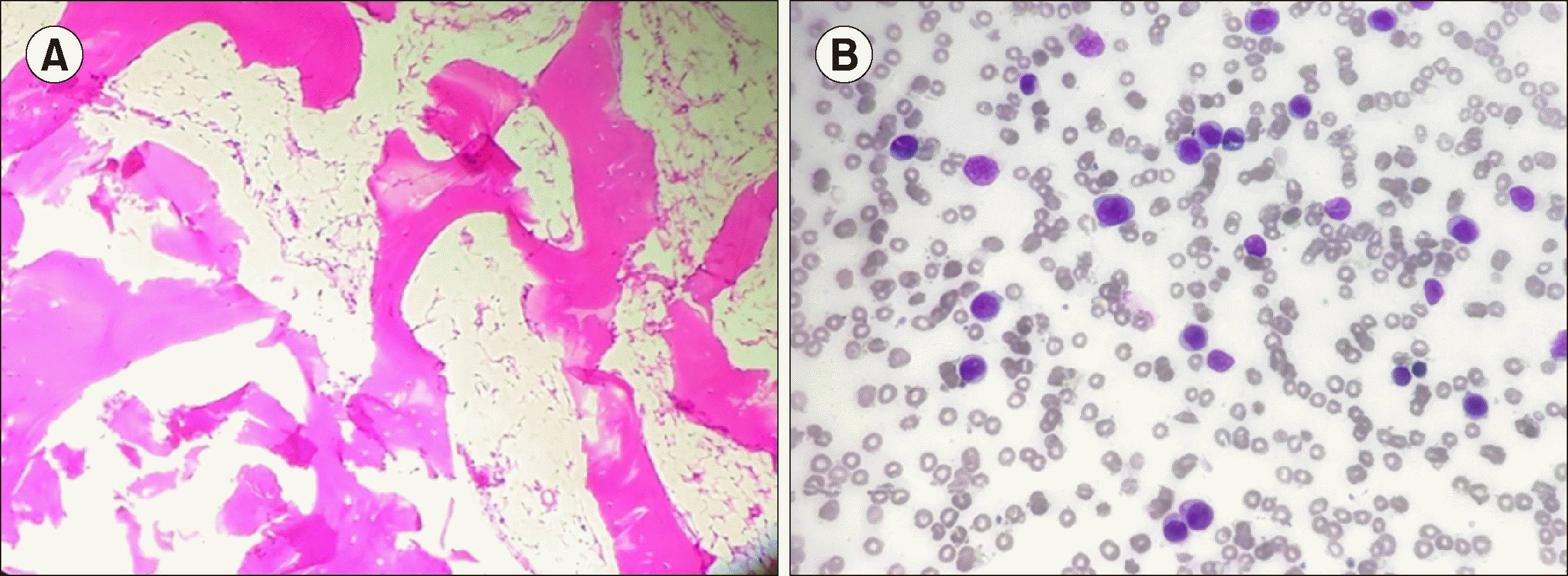
Bone marrow aspirate showed fat-rich marrow fragments with a paucity of normal hematopoietic elements. Biopsy (Fig. 1A) revealed a hypocellular marrow, with overall cellularity of less than 5%. Stress cytogenetic testing showed no difference in sensitivity to mitomycin C between the patient and the control. Peripheral blood smear showed a normal karyotype of 46 XX. PNH testing by multiparametric flow cytometry (FCM) fluorescent aerolysin (FLAER)-based assay (sensitivity 1%) showed absence of PNH clones. A final diagnosis of severe AA was made after ruling out secondary causes of myelosuppression.
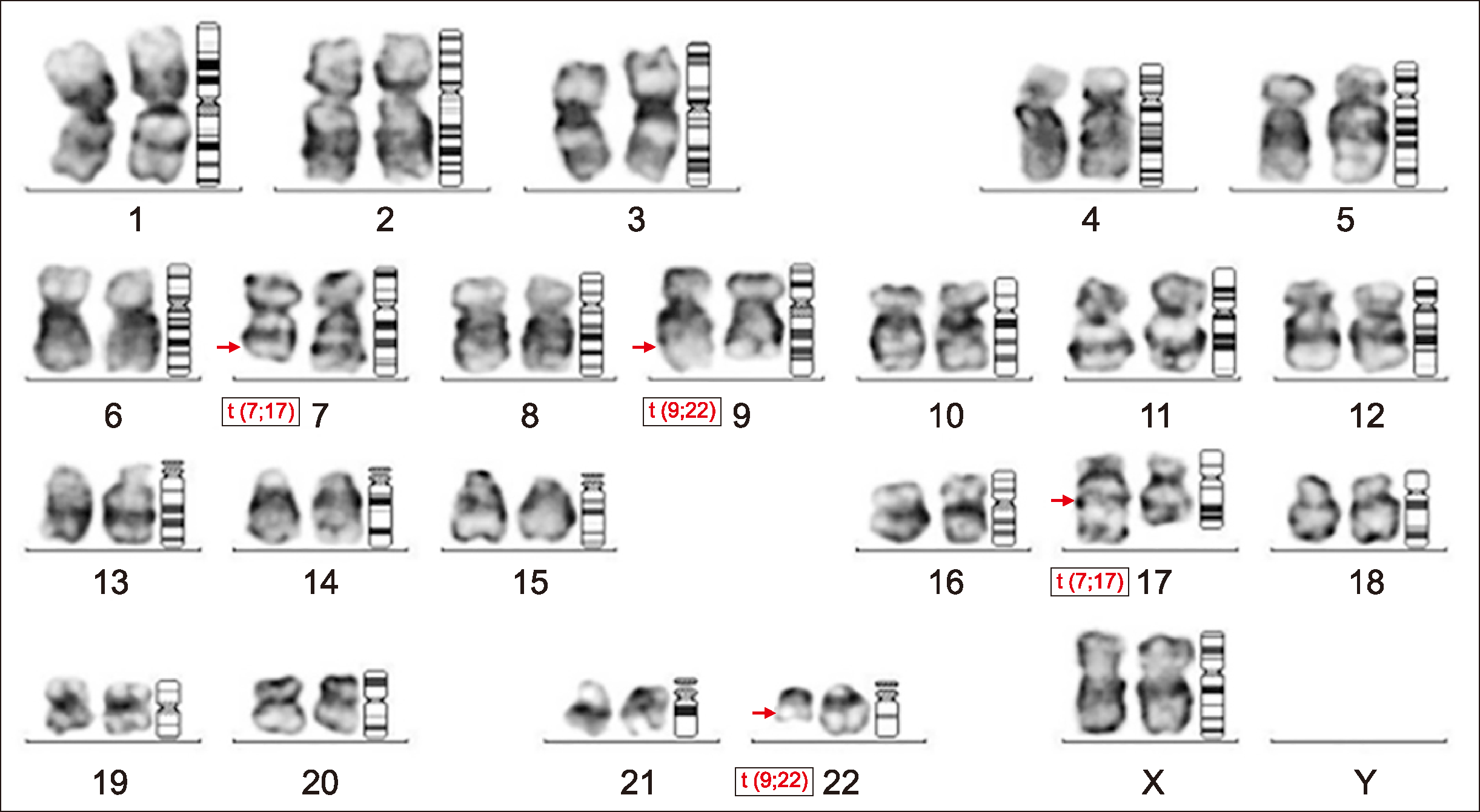
The patient was started on oral IST (cyclosporine and anabolic steroids) and counseled to receive bone marrow transplant as the treatment of choice. Due to resource constraints, the patient received oral immunosuppressive therapy. She was transfusion-independent for 4 years and showed partial response to therapy. The patient again presented to the outpatient department with anemia symptoms in June 2017, with no history suggestive of bleeding or recurrent infection. Examination revealed no organomegaly. A routine workup with PNH assay was ordered. The FCM-FLAER-based assay showed 59% PNH clones in granulocytic series and 47.3% clones in monocytic series. The patient’s cytopenia continued to worsen with no response to treatment. Repeat PNH testing was advised. PNH testing was performed in January 2018 using CD45, FLAER, CD15, CD24, CD64, and CD14 on a multiparametric FCM platform. CD45 side-scatter (Ssc) dot plots showed a cluster of events in CD45 dim and a moderate side-scatter area in the blast region. Peripheral smear examination of the same sample revealed 25% blasts and no basophils. Bone marrow evaluation showed >90% blasts (Fig. 1B), which were positive for non-specific esterase and myeloperoxidase stain on cytochemistry analysis. FCM immunophenotyping of the blasts showed positivity for CD13, CD33, CD34, HLA-DR, MPO, CD64, and CD11c, suggesting AML with monocytic differentiation. Cytogenetic analysis of the bone marrow revealed t (9;22)(q34; q11) and t (7;17)(q22:q21) in all 20 metaphases (Fig. 2). The patient showed no splenomegaly at this presentation. A final diagnosis of clonal evolution to Ph-positive AML in a known case of AA was made. The patient had febrile neutropenia and was treated with broad-spectrum antibiotics and antifungal agents. She denied any chemotherapy and left against medical advice. The patient has since been lost to follow-up.
Discussion
The exhaustion of hematopoietic stem cells by immunological attack leads to pancytopenia in acquired AA. Immunological escape by glucosyl phosphatidylinositol (GPI) anchor or human leukocyte antigen (HLA)-deficient cells gives them a survival advantage. This is a possible explanation for the >50% of AA cases showing clonal hematopoiesis in the form of tiny PNH clones on FCM, chromosome 6p uniparental disomy on single nucleotide polymorphism (SNP) array karyotyping, or positivity for mutations [3]. The detection of PNH clones has clinical utility, as their presence predicts a better response to IST and serves as a surrogate marker to rule out IBMF [2, 7]. Clinically significant diatheses including PNH, MDS, or AML are seen in only 15–25% of acquired AA cases. The exact mechanism for this observation is still not completely understood. The high risk of clonal evolution in IBMF is attributed to differences in the basic pathogenesis of these disorders.
The evolution of AA to AML is very well described [3]. The Philadelphia chromosome in the context of AA is described in rare case reports at baseline or on evolution to ALL or CML in the literature [8, 9]. Philadelphia-positive AML is itself a very rare entity. To the best of our knowledge, this is the first described case of Ph-positive AML in a known case of AA. A review of Ph-positive AML literature showed that many cases evolved in known cases of MDS or harbored additional MDS-defining cytogenetic abnormalities to become AML with myelodysplasia-related changes as per the recent WHO classification [4, 10, 11]. De novo MDS cases harboring Philadelphia chromosome at baseline have also been reported [6]. The significant overlap between AA, PNH, MDS, and AML may explain the Ph positivity at the time of clonal evolution in our case. Our case report described an entire course of disease in one patient, where Ph-positive AML develops on the wheels of PNH in known cases of AA. BCR-ABL is considered a class I mutation in reference to AML and provides proliferation advantage to neoplastic clones [10]. Co-existing cytogenetic and molecular events may determine the preexisting illness in the form of AA, MDS, or de novo leukemia. In our case, the patient refused treatment or molecular workup that might have provided insight into the BCR-ABL transcript type or other coexisting mutations.
The availability of SNP array karyotyping, exome sequencing, and next-generation sequencing in clinical labs has increased the momentum for the search for predictors of clonal evolution in AA. Cytogenetic abnormalities including -7/del 7q are associated with increased risk of progression to MDS/AML [12], while DNMT3A and ASXL1 mutations are good molecular predictors. These factors are also reported at variable frequencies in age-related clonal hematopoiesis. Hence, the application of this information in predicting prognosis and tailoring treatment is still not part of clinical practice. Our interesting case suggests that molecular and genetic events in AA, MDS, and AML are complex and overlapping.
 XML Download
XML Download