

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Hormonal regulation is critical for maintaining metabolic homeostasis in mammals. As a major anabolic hormone, pancreatic insulin is responsible for activating signals in various tissues that promote fuel storage in response to feeding [1]. Insulin counterregulatory hormones, such as glucagon, catecholamine, and cortisol, oppose the action of insulin by promoting fuel mobilization and usage during times of low nutrient intake [2]. Interestingly, among the insulin counterregulatory hormones, glucagon and catecholamine function through the G protein-coupled receptors (GPCRs), which elicit intracellular signaling cascades. In particular, the activation of glucagon and β-adrenergic receptors leads to the activation of GαS, resulting in the activation of adenylate cyclase and an increase in cyclic adenosine monophosphate (cAMP) levels in target cells. cAMP signaling is mainly mediated by protein kinase A (PKA), although some of the downstream signals of cAMP are also mediated by exchange protein directly activated by cAMP (Epac) [3].
In the liver, PKA is responsible for the activation of glucose production by activating both glycogenolysis (via the activation of glycogen phosphorylase kinase and the inactivation of glycogen synthase) and gluconeogenesis (via transcriptional mechanisms) [4]. Furthermore, PKA represses lipid biosynthesis by inhibiting glycolysis and lipogenesis via the inhibition of phosphofructokinase-2, pyruvate kinase, and the transcription factor—sterol regulatory element-binding protein 1c (SREBP-1c)—and cholesterol biosynthesis via the inhibition of 3-hydroxy-3-methyl-glutaryl-CoA (HMG-CoA) reductase. In white adipocytes, PKA triggers the mobilization of stored triglycerides by activating lipolysis, thus releasing free fatty acids from the tissue [5]. Upon activation, PKA directly phosphorylates two major targets, perilipin-1 (PLN1) and hormone-sensitive lipase (HSL). Phosphorylation of PLN1 promotes the release of comparative gene identification-58 (CGI-58), which forms a complex with adipose triglyceride lipase (ATGL). Thus, PKA is critical for activating lipolysis in adipose tissues by modulating two key enzymes in the process, ATGL and HSL. In brown adipocytes, adaptive thermogenesis in response to low temperatures is achieved via the sympathetic nervous system-mediated activation of cAMP/PKA signaling [6]. Activation of PKA also elicits enhanced lipolysis in brown adipocytes in a manner similar to that in white adipocytes. Furthermore, PKA promotes the transcription of nuclear genes encoding key players in mitochondrial metabolism, including uncoupling protein-1 and mitochondrial oxidative phosphorylation protein, via transcriptional activation of peroxisome proliferator-activated receptor γ (PPARγ) coactivator 1 α (PGC-1α). Thus, the action of PKA includes both acute phosphorylation-mediated events as well as chronic transcriptional activation of metabolic enzymes that would affect metabolic flux, to confer the response to these hormones.
cAMP response element-binding protein (CREB) is a major transcription factor that mediates cAMP/PKA-driven transcriptional events in mammals [78]. In response to increased cAMP levels, the catalytic subunit of PKA is released from the regulatory subunit and enters the nucleus. CREB is usually bound to the cAMP response element (CRE) on the target promoter. PKA-mediated phosphorylation of CREB on its serine 133 residue promotes its conformational change, enabling it to interact with its coactivator, CREB binding protein (CBP)/p300, leading to the transcriptional activation of CRE-containing target genes. For example, in the liver, CREB is responsible for the increased transcription of gluconeogenic genes, such as glucose 6 phosphatase (G6Pase) and phosphoenolpyruvate carboxykinase (PEPCK), upon activation of the glucagon-cAMP-PKA signaling axis. Recently, a new class of CREB coactivators, termed CREB-regulated transcription coactivators (CRTCs) have been identified [910]. Among the family members, CRTC2 has emerged as a major mediator of cAMP signaling pathways in metabolic tissues. In this review, we delineate the recent findings regarding the role of CRTC2 in metabolic homeostasis in various tissues, from studies utilizing CRTC2-knockout (KO) mouse models.
Go to : 
IDENTIFICATION OF CRTC PROTEINS
CRTC proteins, also known as transducers of regulated CREB activity (TORCs), were first identified as novel transcriptional coactivators of CREB, by screening an expression library [9]. By using CRE luciferase reporter system, CRTC1, a founding member of the CRTCs, was shown to increase CRE luciferase activity upon cAMP treatment in HEK293 cells. Interestingly, CRTC1 was first reported as an oncogene following a fusion event. The fusion gene, known as MECT1-MAML2, was generated via a recombination event, in which DNA sequences encoding the CREB-binding domain of CRTC1 (termed mucoepidermoid carcinoma translocated-1 [MECT1]) was fused to the C-terminal transcriptional activation domain of the Notch coactivator, mastermind-like 2 (MAML2), leading to the generation of a fusion gene encoding a strong transcriptional coactivator, found in mucoepidermoid carcinoma [11]. The oncogenic potential of the MECT1-MAML2 fusion protein is achieved by the aberrant activation of CREB target genes, since this fusion protein also lacks a DNA-binding domain. By homology screening, two other mammalian paralogues, CRTC2 and CRTC3, were identified in mammals. Furthermore, orthologues of CRTCs have also been identified in other major experimental model organisms, such as Caenorhabditis elegans and Drosophila melanogaster, thus showing an evolutionary conservation and demonstrating the critical role of this CREB coactivator family in cAMP-dependent transcriptional pathways [7].
Go to :
THE STRUCTURE OF CRTC2, AND CRITICAL RESIDUES FOR ITS REGULATION
In spite of their importance in the cAMP-dependent transcriptional events, the structure of CRTC proteins has not been reported to date and only a rough description of critical residues is available [7]. The amino terminus of CRTC proteins is termed the CREB-binding domain (CBD) due to its importance in binding to CREB and other basic leucine zipper (bZIP)-type proteins. The carboxy terminus is regarded as a transactivation domain (TAD), which interacts with various basic transcriptional machineries as well as with other transcriptional coactivators. The central part of this protein is generally regarded as a regulatory domain, which contains many serine/proline amino acids and is a target for various post-translational modifying enzymes that will be described in more detail below. A recent study has provided detailed structural insights into the role of the amino terminal CBD of CRTC2 [12]. While CRTC2 CBD lacks the intrinsic DNA-binding affinity due to the paucity of basic amino acid residues, the amino terminal CBD forms a 28-residue alpha helix that interacts not only with CREB bZIP residues, but also with the phosphate groups of CRE-containing DNA sequences in a nonspecific manner. Thus, it is predicted that CRTC2 may stabilize the CREB/DNA complex on the promoter and increase the specific binding of CREB onto the target promoter. We have recently shown that the CBD of CRTC2 is a target of protein arginine methyltransferase 6 (PRMT6) [13]. Upon its activation and nuclear entry, CRTC2 is targeted by PRMT6. Four evolutionarily conserved arginine residues of CRTC2 (arginine 51, 99, 120, and 123) in the CBD are asymmetrically dimethylated, resulting in enhanced interaction between CRTC2 and CREB on the gluconeogenic promoters.
The central domain of CRTC2 consists of serine/proline-rich sequences, and is considered as a regulatory domain for the protein. Serine 171 residue (in the murine CRTC2) is regarded as a major site for the regulation of the protein. In the absence of cAMP signaling, serine 171 residue of CRTC2 is phosphorylated via AMP-activated protein kinase (AMPK) and AMPK-related kinases, such as salt-inducible kinases (SIKs), and the protein is found mainly in the cytosol, and associated with 14-3-3 [1014]. Upon activation of cAMP signaling, PKA-mediated phosphorylation of AMPK and its related kinases inhibits their activity. In addition, serine/threonine phosphatases, such as protein phosphatase 4 (PP4) and protein phosphatase 2B (PP2B), have been shown to be responsible for the dephosphorylation of CRTC2 at serine 171 residue, leading to the nuclear localization and activation of this coactivator [1516]. Interestingly, PP4 and PP2B have been shown to be activated in a cAMP-dependent manner. While the expression of regulatory subunit 3 of PP4 (PP4R3) is induced in response to cAMP signaling, cAMP-dependent induction of the inositol-1,4,5-trisphosphate receptor leads to increased intracellular calcium levels and the resultant activation of PP2B. The serine 171 residue is also a target of O-linked N-acetylglucosamine (O-GlcNAc) transferase (OGT) [17]. The O-GlcNAcylation of serine 171 residue of CRTC2 is increased under conditions of chronic exposure to glucose such as in type 2 diabetes mellitus (T2DM), which prevents its phosphorylation and inactivation in the liver. Thus, O-GlcNAcylation of CRTC2 is considered to be a potential mechanism for the increased hepatic gluconeogenesis in diabetes.
The carboxy terminal TAD domain has also been studied recently. The lysine 628 residue of CRTC2 might be critical for its transactivation potential. CREB and CRTC2 form a stable complex on CRE-containing promoter sequences, in conjunction with another CREB coactivator, CBP/p300. Indeed CBP/p300-mediated acetylation of CRTC2 at lysine 628 promotes a tighter binding of the active CREB-CRTC2-CBP/p300 complex with the promoter, while sirtuin 1 (Sirt1)-dependent deacetylation inhibits the binding [18]. The same lysine 628 is also a target of polyubiquitination. Constitutively photomorphogenic 1 (COPI) ubiquitin ligase has been shown to be responsible for this event, which leads to the degradation of CRTC2 via the proteasome machinery. Finally, the TAD of CRTC2 has also been shown to be critical in recruiting the basal transcription factor that is responsible for histone modification. The TAD of CRTC2 directly binds to the histone acetyltransferase, K(lysine) acetyltransferase 2B (KAT2B), which leads to the increased histone 3 lysine 9 acetylation of gluconeogenic promoters and directs the activation of the transcriptional program in response to cAMP [19].
In summary, the role of each domain in CRTC2 has been delineated in relation to the other regulatory proteins in the various pathways (Fig. 1). The relevance of the specific residues in the disease state should be addressed in the in vivo knock-in mouse models bearing specific mutations in the future.
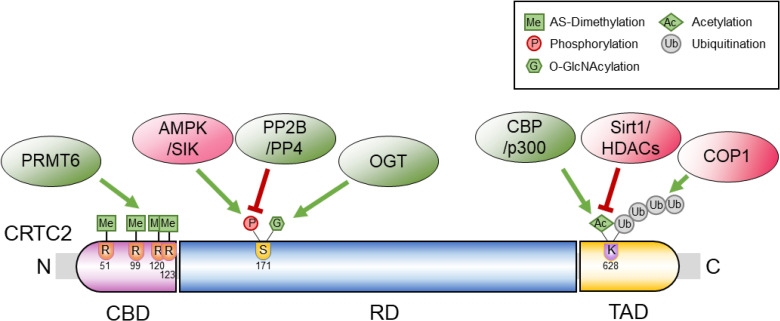 | Fig. 1The structure and regulatory mechanisms of CREB-regulated transcription coactivator 2 (CRTC2). CRTC2 comprises a CREB-binding domain (CBD), a regulatory domain (RD), and a transactivation domain (TAD), each of which is targeted by various regulatory enzymes described in the text. CREB, cyclic adenosine monophosphate (cAMP) response element-binding protein; PRMT6, protein arginine methyltransferase 6; AMPK, adenosine monophosphate-activated protein kinase; SIK, salt-inducible kinase; PP2B, protein phosphatase 2B; PP4, protein phosphatase 4; OGT, O-linked β-N-acetylglucosamine transferase; CBP, CREB-binding protein; Sirt1, sirtuin 1; HDAC, histone deacetylase; COP1, constitutive photomorphogenic 1; N, amino-terminus; R, arginine; S, serine; K, lysine; C, carboxy-terminus.
|
Go to :
THE ROLE OF CRTC2 IN THE CONTROL OF METABOLIC PATHWAYS
Liver
Glucose metabolism
As a major CRTC protein in the liver, the role of CRTC2 in hepatic glucose metabolism has been extensively studied. Under fasting conditions, glucagon-mediated activation of CRTC2 is crucial in activating CREB target genes involved in gluconeogenesis, such as PEPCK and G6Pase, leading to increased gluconeogenic flux in the liver [14]. In addition, the CREB-CRTC2-dependent transcriptional machinery is crucial for the transcriptional regulation of hepatic gluconeogenesis. PGC-1α was first identified as a coactivator of PPARγ in brown adipocytes and it is regarded as a crucial regulator of hepatic gluconeogenesis via interaction with nuclear receptors, including glucocorticoid receptor (GR) or hepatic nuclear factor 4 (HNF4) α, as well as the forkhead box protein O1 (FoxO1) [20]. Indeed, CRTC2 activates the transcription of PGC-1α in conjunction with CREB. Thus, CRTC2 can enhance the hepatic gluconeogenic program, both directly (via association with CREB) and indirectly (via the increased expression of PGC-1α) [14]. Estrogen-related receptor gamma (ERRγ) is another transcriptional target of CREB/CRTC2 that is involved in the glucose metabolism in the liver [21]. Expression of ERRγ is elevated in response to cAMP signaling, in a CREB/CRTC2-dependent manner. Elevation of ERRγ expression has been observed in mouse models of T2DM and the inhibition of ERRγ activity by treatment with the inverse agonist, GSK5182, restores euglycemia; thus showing that ERRγ might be a potential target for a novel T2DM therapeutics. These studies show that CRTC2 is a crucial coordinator of hepatic gluconeogenesis via controlling multiple transcriptional machineries in the liver.
The crucial role of CRTC2 in the control of hepatic gluconeogenesis has been verified in vivo by using several mouse models. Acute inhibition of CRTC2 activity by RNA interference reduces blood glucose levels as a result of reduction in hepatic gluconeogenesis [1422]. Interestingly, contradictory findings have been reported by two groups utilizing distinct systemic CRTC2-KO mice. In the first study, systemic CRTC2-KO mice were generated by the deletion of exon 4 through 11 of CRTC2, resulting in the fortuitous production of an in-frame mutant of the CRTC2 protein that lacks the regulatory domain, including the crucial serine 171 residue [23]. The resultant in-frame mutant functions as a constitutively active coactivator of CREB-dependent transcription in cultured cells [7]. While the resultant CRTC2-KO mice did not exhibit alterations in glucose metabolism compared with the wild-type mice, this result needs more stringent confirmation due to the problematic strategy in generating the KO mice. In the second study, exon 1 of CRTC2 was targeted during the generation of systemic CRTC2-KO mice [24]. As a result, no functional CRTC2 gene product was produced. Unlike in the previous studies, CRTC2-KO mice lacking the entire CRTC2 protein displayed reduced blood glucose levels and improved glucose tolerance compared with their wild-type littermates, which is consistent with the results of earlier acute knockdown studies. Indeed, we were able to confirm the results of the latter study by using liver-specific CRTC2-KO mice, and showing that CRTC2 is a crucial regulator of glucose metabolism by controlling hepatic gluconeogenesis in vivo [25].
Lipid metabolism
Recent studies have revealed the role of CRTC2 in hepatic lipid metabolism by utilizing systemic CRTC2-KO mice. Wang and colleagues showed that CRTC2 regulates hepatic lipogenesis via a non-genic manner [26]. They showed that under fasting conditions, CRTC2 is tightly associated with Sec31A, a component of the coat protein complex II (COPII) complex, thus inactivating COPII activity. Upon feeding, mammalian target of rapamycin (mTOR) complex 1 phosphorylates serine 136 residue of CRTC2, thus releasing Sec31A from this factor. Sec31A, in turn, interacts with Sec23, forming an active COPII complex, leading to the translocation and subsequent proteolytic cleavage-dependent activation of the SREBP-1c. The authors further showed that enhanced phosphorylation of CRTC2, via mTOR complex 1, accounts for the increased hepatic lipogenesis seen in obesity. In addition, another recent study suggested a role of CRTC2 in controlling hepatic cholesterol metabolism. The authors demonstrated that CRTC2 is essential for activating the transcription of SREBP-2, a master regulator of cholesterol metabolism, via co-activation of CREB and inhibition of FoxO1 [27]. Furthermore, in conjunction with SREBP-2, CREB/CRTC2 has also been shown to enhance the transcription of HMG-CoA reductase, thus suggesting a critical role of CRTC2 in controlling hepatic cholesterol homeostasis. While these studies provided intricate mechanistic roles of CRTC2 in regulating hepatic lipid metabolism, liver-specific CRTC2-KO mouse models need to be utilized to confirm these findings that have been identified in studies that mainly used whole-body CRTC2-KO mice.
We have explored the role of CRTC2 in hepatic lipid metabolism by using liver-specific CRTC2-KO mice [25]. Unlike previous reports that used whole-body CRTC2-KO mice, we found that liver-specific depletion of CRTC2 ameliorates lipid accumulation in the liver under conditions of diet-induced obesity (DIO). Depletion of CRTC2 specifically reduces the expression of miR-34a, a microRNA that is known to be induced in obesity in humans, leading to the induction of its molecular targets, Sirt1 and PPARα, in the liver. Activation of Sirt1 and PPARα not only reduces lipid accumulation, by inducting fatty acid β oxidation, but also enhances whole-body energy metabolism via the induction of fibroblast growth factor 21 (FGF21). Interestingly, while we observed a reduction in miR-34a upon liver-specific depletion of CRTC2, under both fasting and feeding conditions in DIO mice, the induction of the Sirt1/PPARα/FGF21 axis was mainly observed under fasting conditions. Identification of potential targets of miR-34a in the feeding state might be necessary to fully understand the molecular events that lead to the observed improvement in metabolic homeostasis upon liver-specific depletion of CRTC2.
Pancreatic β-cells
In pancreatic β-cells, CREB is activated in response to feeding (via glucose and incretin hormones, such as glucagon-like peptide 1 [GLP-1] and gastric inhibitory polypeptide), in part via the dephosphorylation and activation of CRTC2. While glucose metabolism leads to the induction of intracellular calcium influx, leading to the activation of protein phosphatase 2B (PP2B) (also known as calcineurin)-mediated dephosphorylation and activation of CRTC2, incretin-dependent signaling increases cAMP signaling cascades in the β-cells, thus activating CRTC2 by inhibiting SIKs [10]. The activation of CREB/CRTC2 in β-cells is essential for maintaining the function and survival of these cells, in part via the regulation of insulin receptor substrate 2 (IRS2) [28]. Indeed, the importance of CRTC2 in β-cell function has been verified in a study that utilized β-cell-specific CRTC2-KO mice [29]. β-Cell-specific CRTC2-KO mice displayed oral glucose intolerance due to a reduction in insulin secretion in response to the oral glucose gavage. Unlike in the previous observation, CRTC2-KO islets retained the ability to secrete insulin in response to high glucose concentrations ex vivo. However, the depletion of CRTC2 in β-cells significantly impaired incretin-mediated insulin secretion in the cultured islets, showing that the incretin-cAMP signaling pathway is more prominent in regulating CRTC2-dependent transcriptional events in β-cells. In addition to its role in the regulation of β-cell survival via the induction of IRS2, CREB/CRTC2 has also been shown to maintain β-cell function via the induction of V-maf musculoaponeurotic fibrosarcoma oncogene homolog A (MafA), a β-cell-specific transcription factor that is critical in maintaining insulin gene expression. Chronic exposure to glucose reduced incretin-PKA signaling by activating the mTOR-hypoxia-inducible factor-1 α (HIF-1α)-PKIβ pathway, leading to the inhibition of CRTC2-dependent transcriptional events and the subsequent deterioration of β-cell function. In a latter study, the authors utilized a mouse insulin promoter (MIP)-CreERT system for the temporal KO of CRTC2 expression in β-cells in adult mice. It will be interesting to explore the role of CRTC2 in β-cell development and/or maturation by using constitutive Cre drivers to generate β-cell-specific CRTC2-KO mice in future studies.
Small intestinal L cells
Among the endocrine cell types in the small intestine, L cells are the predominant secretors of GLP-1, a major incretin that regulates glucose homeostasis [3031]. In the intestinal L cells, GLP-1 is initially produced as a precursor protein, proglucagon that is encoded by glucagon (Gcg) gene. Maturation of proglucagon to the active GLP-1 protein requires the action of a specific prohormone convertase (PC)1/3 in L cells. In pancreatic α cells, the predominant expression of PC2 leads to the generation of glucagon, instead of GLP-1, thus generating cell-type specific hormones from the same precursor protein. GLP-1 release from the intestinal L cells in response to feeding, has been shown to be enhanced by various fuels that are present in the intestinal lumen. The entry of glucose, via sodium-dependent glucose transporter 1 (SGLT1) or glucose transporter 2 (GLUT2), leads to the activation of glucose oxidation and the generation of adenosine triphosphate (ATP), leading to increased calcium influx into the L cells. In addition, free fatty acids or bile acids can activate GPCRs, such as GPR40/120 for fatty acids and G protein-coupled bile acid receptor 1 (GPBAR1, also known as Takeda-G-protein-receptor-5 [TGR5]) for bile acids, leading to the activation of the cAMP signaling pathway [323334]. Both calcium-dependent signaling and cAMP signaling are required for the exocytosis of GLP-1-containing granules in the L cells. In our recent study, we demonstrated that CRTC2 is critical for the regulation of GLP-1 secretion from L cells [35]. Knockdown of CRTC2 reduces bile acids and cAMP-dependent GLP-1 secretion, whereas the constitutive activation of CRTC2 pathway promotes increased exocytosis of GLP-1 in cultured L cells. Interestingly, the CREB/CRTC2-dependent transcription pathway plays an essential role in all aspects of GLP-1 production and secretion. Using RNA-seq analysis, we found that the depletion of CRTC2 in L cells led to the reduced expression of proglucagon and PC1/3, thus explaining the reduced production and maturation of GLP-1. Furthermore, depletion of CRTC2 led to the reduced expression of PGC-1α, resulting in a reduced capacity of mitochondrial oxidative phosphorylation and a subsequent reduction in calcium influx into the cells. Indeed, intestine-specific KO of CRTC2 in mice results in reduced plasma GLP-1 levels, thereby leading to glucose intolerance due to impaired β-cell function and maintenance in the absence of the critical incretin hormone. This study demonstrated that the CRTC2-dependent transcriptional pathway is essential for maintaining glucose homeostasis via its regulation of GLP-1 secretion in the intestinal L cells. It will be of interest to compare/contrast the relative contributions of CRTC2 in β-cells and intestinal L cells in the control of glucose metabolism in future studies.
Adipose tissues
Since CRTC3 is the major CRTC isoform expressed in adipose tissues, the function of CRTC3, rather than CRTC2, has been predominantly studied in adipose tissues (see the following sections for more information). In a study that utilized transgenic mice expressing constitutively active SIK2 in brown adipocytes, inactivation of CRTC2 in brown adipocytes led to increased body weight and a decrease in the cold temperature-associated elevation of body temperature, in part via the reduction in the levels of uncoupling protein 1 and PGC-1α [36]. In white adipocytes, CREB has been shown to be activated in obesity and to be critical in instigating insulin resistance [37]. CREB increases the expression of the transcriptional repressor, activating transcription factor 3 (ATF3), thereby leading to the reduced expression of adiponectin and GLUT4 in adipocytes. The role of CREB in instigating insulin resistance has been confirmed in adipocyte-specific transgenic mice expressing a dominant negative form of CREB (F-ACREB mice). Compared with control mice, F-ACREB mice show increased whole-body insulin sensitivity under diet-induced or genetic obesity. Conversely, SIK2-KO mice display increased body fat and impaired insulin sensitivity under DIO [38]. In SIK2-KO adipocytes, constitutive activation of CRTC2 has been observed, which results in the induction of ATF3 and the subsequent downregulation of adiponectin and GLUT4 expression. In CRTC2-KO primary adipocytes, we confirmed that the depletion of CRTC2 enhanced insulin signaling via restoring the expression of adiponectin and GLUT4, thus accentuating the importance of this coactivator in adipose tissue. However, further studies are necessary in adipocyte-specific CRTC2-KO mice, to explore the role of CRTC2 in adipocytes.
Go to :
CRTC2 AS A TRANSCRIPTIONAL COACTIVATOR FOR OTHER bZIP TRANSCRIPTION FACTORS
Since CRTC2 possesses a domain that interacts with the bZIP domain, it was reasonable to assume that it could interact with other bZIP transcription factors belonging to the CREB family. Indeed, it has been shown that CRTC2 interacts with ATF6, a class of endoplasmic reticulum (ER)-bound bZIP transcription factors, in the liver [39]. ATF6 is regarded as one of three critical factors that are important for triggering the unfolded protein response (UPR) pathway. ATF6 is initially generated as an ER membrane-bound protein and is anchored in the membrane of the rough ER in the normal cellular state. Upon ER stress, either by the production of excessive unfolded proteins or by nutritional stress, ATF6 relocates to the Golgi apparatus and is cleaved to generate an active transcription factor moiety that enters the nucleus. Interestingly, ER stress also enhances the release of ER-stored calcium into the cytosol, thereby leading to the activation of PP2B and the subsequent activation of CRTC2 by dephosphorylation of serine 171 residue. The concomitant activation of ATF6 and CRTC2 thus provides a mechanism for the enhanced expression of UPR pathway genes in the liver. We have also shown that CRTC2 acts as a coactivator for another ER-bound bZIP transcription factor, CREBH (also known as CREB3L3). CREBH is a member of the CREB3 transcription factor family [40]. Although its proteolytic cleavage is not regulated by the nutrient status, the formation of the active form of nuclear CREBH is enhanced under fasting conditions, due to the increased expression of CREBH. Furthermore, unique CREBH binding sites that are distinct from CREB binding sites, have been identified in the promoter of PEPCK and G6Pase, indicating that CRTC2-dependent activation of hepatic gluconeogenesis requires both CREB and CREBH. Further studies are needed to identify the potential interacting partners of CRTC2 among the other bZIP transcription factors.
Go to :
FUNCTIONS OF OTHER CRTC PROTEINS: HOMOLOGUES AND PARALOGUES OF CRTC2
CRTC1 is predominantly expressed in the central nervous system. In the initial study that utilized CRTC1 systemic KO mice, depletion of CRTC1 in the hypothalamic arcuate cells reduced the production of cocaine- and amphetamine-regulated transcript (CART), an anorexigenic protein, thereby leading to the dysregulation of feeding behavior. As a result, CRTC1-KO mice exhibited obesity, in part due to leptin resistance [41]. Subsequent studies showed that CRTC1 is expressed in various cell types in the brain, where it functions as a regulator of distinct phenomenon, such as the circadian rhythm (suprachiasmatic nucleus), long-term memory (hippocampus), and mood disorders (hippocampus; for further information see [42]). However, abundant expression of CRTC3 is observed in both white and brown adipocytes. The function of CRTC3 in the control of energy expenditure has been delineated by using CRTC3-KO mice. As stated previously, the β-adrenergic receptor-Gαs-cAMP signaling axis is critical for instigating lipolysis in white adipocytes and adaptive thermogenesis in brown adipocytes. Systemic CRTC3-KO mice are resistant to DIO and insulin resistance, due to the reduced expression of regulator of G protein signaling 2 (RGS2), a negative regulator of G protein activity [43]. In addition, brown adipocyte-specific KO of CRTC3 in mice results in reduced adiposity and improved cold tolerance. This effect is, in part, mediated by the reduced expression of miR-206 in brown adipocytes, thus promoting sympathetic nerve innervation and vascularization in brown adipose tissues (cAMP-inducible coactivator CRTC3 attenuates brown adipose tissue thermogenesis) [44]. These results highlight the importance of CRTC2 paralogues in the regulation of metabolic pathways in mammals.
CRTC orthologues in lower eukaryotes have also been shown to be critical for the regulation of energy metabolism. Only a single orthologue of CRTC, termed dTORC, has been identified in the fruit fly, D. melanogaster. As is the case for CRTC1 in mice, dTORC is mainly expressed in the brain and has been shown to regulate energy balance as well as resistance to oxidative stress [45]. dTORC-mutant flies show a reduced longevity, due in part to their reduced potential to store fuels as triglycerides and glycogen and an impaired response to oxidative stress. As in the case of the mammalian orthologues, dTORC activity is regulated by a conserved serine residue in the regulatory domain, which is targeted by the fly orthologue of SIK. C. elegans, another widely utilized model organism in metabolic research, also possesses a sole orthologue of CRTC, CRTC-1. In this organism, CRTC-1 also serves as a transcriptional coactivator for the CREB orthologue, C. elegans homolog of mammalian cAMP response element binding protein 1 (CRH-1). In contrast to fly CRTC (dTORC), worm CRTC (CRTC-1) decreases longevity. Surprisingly, deletion of either CRTC-1 or CRH-1 promotes oxidative resistance and the resultant increase in longevity in C. elegans [46]. Thus, further studies are necessary to discern the distinctive functions of dTORC and CRTC-1 in the respective organisms, including their roles in the regulation of tissue-specific metabolic pathways.
Go to :
CONCLUSIONS
Since the identification of CRTC proteins in 2003, the role of CRTC2 in metabolic tissues has been extensively studied. Here, we reviewed the current literature regarding the regulatory mechanisms of CRTC2, as well as its function in the control of metabolic pathways. The GPCR-mediated elevation of cAMP signaling promotes the activation of CRTC2 via the inhibition of the AMPK family of kinases and the activation of PP2B/PP4 phosphatases, PRMT6, and the coactivator, CBP/p300. In the liver, CRTC2 regulates hepatic gluconeogenesis by co-activation of the CREB/CREBH pathway as well as through the induction of transcriptional regulators, including PGC-1α and ERRγ. While CRTC2 may inhibit lipogenesis and promote cholesterol biosynthesis in certain systems, tissue-specific depletion of CRTC2 in the liver reduces DIO-induced fatty liver and insulin resistance. CRTC2 has been shown to be critical for maintaining β-cell function by directly controlling glucose-stimulated insulin secretion and the proliferation of pancreatic β-cells, as well as by controlling the production of mature GLP-1 from the intestinal L cells. In white adipose tissues, increased CRTC2 activity may be a cause of the pronounced insulin resistance and impaired glucose metabolism seen under DIO conditions (see Fig. 2 for more complete information). Further studies using other types of tissue-specific CRTC2-KO mice are necessary to provide more information regarding the role of CRTC2 in the control of metabolic homeostasis in a cell and tissue-specific manner. More importantly, more rigorous collaborative studies are necessary to confirm whether the current findings from mouse models can be applied to humans.
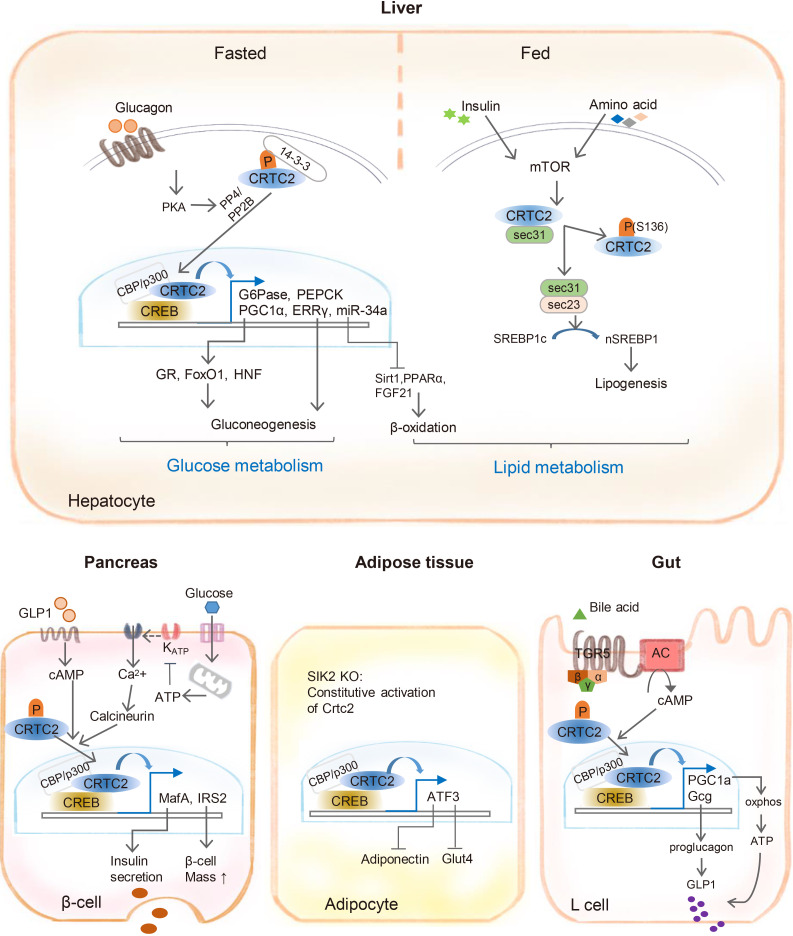 | Fig. 2Role of CREB-regulated transcription coactivator 2 (CRTC2) in the regulation of metabolic pathways in various tissues. In the liver, CRTC2 is critical for the regulation of gluconeogenesis by coactivating cyclic adenosine monophosphate (cAMP) response element-binding protein (CREB) and CREBH. CRTC2 is also involved in the regulation of lipid metabolism via miR-34a-mediated repression of fatty acid β oxidation and the inhibition of sterol regulatory element-binding protein 1 (SREBP-1)-dependent lipogenic gene transcription. CRTC2 is also crucial for the control of systemic glucose homeostasis, by regulating pancreatic β-cell function and the production of glucagon-like peptide 1 (GLP-1) from intestinal L cells. The role of CRTC2 in white adipocytes requires further study, but the depletion of CRTC2 leads to improved glucose uptake as well as an increase in adiponectin secretion by the white adipocytes in vitro. PKA, protein kinase A; PP4, protein phosphatase 4; PP2B, protein phosphatase 2B; CBP, CREB binding protein; PEPCK, phosphoenolpyruvate carboxykinase; ERRγ, estrogen-related receptor γ; GR, glucocorticoid receptor; FoxO1, forkhead box protein O1; HNF, hepatic nuclear factor; Sirt1, sirtuin 1; PPARα, peroxisome proliferator-activated receptor α; FGF21, fibroblast growth factor 21; mTOR, mammalian target of rapamycin; ATP, adenosine triphosphate; IRS2, insulin receptor substrate 2; SIK2, salt-inducible kinase 2; KO, knockout; ATF3, activating transcription factor 3; Glut4, glucose transporter 4; TGR5, Takeda-G-protein-receptor-5; AC, adenylyl cyclase; PGC-1α, peroxisome proliferator-activated receptor gamma coactivator 1α; Gcg, glucagon; oxphos, oxidative phosphorylation.
|
Go to :
 XML Download
XML Download