

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
Monogenic diabetes is a rare form of diabetes specifically caused by a single gene mutation. It includes maturity-onset diabetes of the young (MODY), neonatal diabetes, maternally inherited diabetes and deafness (MIDD), and so on. The genes causing monogenic diabetes play an important role in the development or function of pancreatic β-cells, with some exceptions of genes involved in obesity or insulin resistance. Patients with monogenic diabetes are characterized by early-onset diabetes and a family history of diabetes in multiple first-degree relatives. Monogenic diabetes is frequently misdiagnosed as either type 1 diabetes mellitus (T1DM) or type 2 diabetes mellitus (T2DM). Accurate molecular genetic diagnosis of monogenic diabetes is crucial because the therapeutic approach can be directed by the causative gene in some cases and it helps to identify affected family members.
Among the different types of monogenic diabetes, MODY is the most common form. It is a clinically heterogeneous group of disorders characterized by β-cell dysfunction, causing earlyonset diabetes with autosomal dominant inheritance. Genetic mutations associated with MODY have been reported in 14 genes, to date, as shown in Table 1 [1,2]. There has been a continuous effort to identify and characterize patients with MODY in Korea since 2001. However, some limitations faced were methods for genetic testing, lack of experienced labs, and no referral system focusing on monogenic diabetes. Consequently, it has been difficult to estimate the prevalence of MODY in Korean patients with diabetes and what the most common form of MODY is.
Table 1.
Related genes and associated clinical characteristics and treatments of MODYs
![]()
Over the past decade, there has been a substantial advancement in genome sequencing technology. Next generation sequencing (NGS) based whole exome or targeted panel sequencing is now widely used not only for research purposes but also for the clinical molecular diagnosis of monogenic diabetes. A few recent reports on monogenic diabetes in Korea also rely on these methods. Compared to the conventional Sanger sequencing, targeted panel sequencing has higher sensitivity with higher sequencing depth and improved throughput with the ability to sequence hundreds of genes simultaneously. The number of cases with genetically confirmed monogenic diabetes is increasing and a new era of diagnosis and treatment of monogenic diabetes has emerged. However, clinicians still question the criteria for genetic testing and the interpretation of the sequencing results. It is also not known whether there are different characteristics of monogenic diabetes in East Asians compared to other ethnicities.
In this review, we discuss the latest updates on monogenic diabetes, primarily focusing on MODY in the Korean population. In addition, we suggest an algorithm that could help clinicians diagnose different types of monogenic diabetes based on clinical clues. Finally, genetic tests and non-genetic markers for the accurate diagnosis of monogenic diabetes are discussed.
Go to : 
PREVALENCE AND SUBTYPE FREQUENCY OF MONOGENIC DIABETES IN DIFFERENT ETHNICITIES
Since MODY was first described by Tattersall [3] in 1974, several advanced genetic studies have been conducted. A majority of the large-scale genetic studies were performed in Europeans. In two German studies which involved 40,927 and 2,064 patients who were diagnosed with diabetes before the age of 25 years and had been treated successfully with diet changes or oral drugs for 5 years, the prevalence of MODY was estimated to be 0.14% and 1.8%, respectively [4,5]. In another German/Austrian cohort of 40,757 patients <20 years of age, 0.83% was identified as MODY by clinical criteria [6]. According to the largest and most comprehensive study in the UK in which genetic tests for monogenic diabetes were used [7], the prevalence of MODY was estimated to be at least 1.08%. In the study, the genetic diagnosis rate was 27% (567/2,072), and hepatocyte nuclear factor 1α (HNF1A) (52%) and glucokinase (GCK) (32%) were the most common mutations followed by HNF4α (HMF4A; 10%) and HNF1β (HNF1B; 6%). A recently published report based on a nationwide population-based registry of Norway showed that a total of 6.5% of autoantibody-negative child patients had genetic variants of MODY confirmed by NGS [8].
In comparison to studies on European population, the studies establishing the prevalence and genetic diagnosis rate of monogenic diabetes in Asian population are quite few [9-12]. In a Chinese study, the prevalence of HNF1A- and GCK-MODY in 146 unrelated MODY families was 9% and 1%, respectively [9]. A recent study on 82 autoantibody-negative Chinese patients who were clinically diagnosed with T1DM, showed that 22% had mutations related to monogenic diabetes and up to 6% of Chinese have monogenic diabetes [12]. HNF1A was the most common form (6/18, 33%), however, there were a number of recessive mutations of Wolfram syndrome 1 (WFS1) [12]. An analysis of South Asians in the UK revealed that the genetic diagnosis rate was 12.6% [10]. Among 263 Japanese patients with suspected MODY, 39.2% had one of the four MODY gene mutations—GCK, HNF1A, HNF4A, and HNF1B— when tested with Sanger sequencing, and 21.6% had a GCK mutation [13]. Molecular diagnosis rate in Koreans was 21.1% for patients with clinically suspected monogenic diabetes and 1.1% for patients with overall non-T1DM in the recent largest study [14]. The most common form of MODY was GCK (50%), followed by HNF1A (21.4%) and HNF4A (21.4%).
It is not clear whether MODY prevalence and its subtype frequency differ in ethnic groups. Most of the large-scale studies were conducted in Europeans, hence further studies on different populations are needed. In addition, population-based screening using uniform criteria or universal screening is required to compare prevalence or subtype frequency between different populations. Clinical criteria to screen patients for genetic testing may affect not only the MODY subtype frequency [15] but also the sensitivity in differentiating MODY from early-onset T2DM [10]. As Korean patients with diabetes have lower body mass index (BMI) at diagnosis of diabetes and tend to have earlier onset of diabetes compared to Europeans [16,17], these factors should be considered when interpreting the results. In this context, recent Korean studies suggest BMI ≥27.5 kg/m2 as a threshold for exclusion for genetic tests because none of the participants with BMI ≥27.5 kg/m2 had suggestive pathogenic variants for MODY [14], even though a cut-off based on diagnostic performance was not discerned.
The MODY subtype frequency also varies among studies on the same ethnic group. This is because the results are influenced by the study designs, such as cut-off age determined, genetic testing method used, and whether subjects are limited to cases of strong suspicion or include all cases of early-onset diabetes. In addition, the clinical suspicion rate is relatively low in some subtypes when it is asymptomatic or due to its benign nature as in the case of GCK-MODY. Therefore, an accurate assessment of prevalence and subtype frequency is possible only if diagnosis using genetic and non-genetic tests is performed on a large-scale population based on sufficient suspicion of monogenic diabetes.
Go to :
KOREAN STUDIES FOR MONOGENIC DIABETES
In Korea, the first genetic study on MODY was conducted in 2001. It focused on the HNF1A gene, however, there was only one silent mutation among 69 early-onset T2DM cases (1/69, 1.5%) [18]. During the period of 2003 to 2008, there have been several approaches for the detection of HNF1A mutations in clinically suspected MODY patients and early-onset diabetes. Kim et al. [19] detected a R263L missense mutation using direct sequencing in one patient among 16 early-onset T2DM cases (1/16, 6.3%). Their functional studies revealed that the mutation was associated with decreased insulin production and defective glucose sensing [19]. In one study on 17 children with T2DM and their families, a mutation of the promoter of HNF1A (1/22, 4.5%) was found in a child’s mother, who was diagnosed with diabetes as an adult [20]. In other studies, around 5% of the population was detected to have genetic mutations in HNF1A; 5.0% (2/40) of 23 MODY and 17 early-onset T2DM cases using direct sequencing [21], 4.0% (1/25) of 25 early-onset T2DM cases using DNA chip [22] and 5.2% (5/96) of 96 gestational diabetes mellitus (GDM) cases using direct sequencing [23]. Other MODY mutations were reported less frequently on using direct sequencing; one mutation of GCK [21], HNF4A [21], and HNF1B [24], respectively.
Since 2015, studies using whole exome sequencing (WES) have been conducted on Korean subjects. Shim et al. [25] did not find any known disease-causing alleles of MODY; however, they reported three candidate gene variants in PTPRD, SYT9, and WFS1 found in six MODY probands and their families. In another early-onset diabetes population, four pathogenic or likely pathogenic missense variants in HNF4A, HNF1A, and ABCC8 were found using WES [26]. In the case of WES for three patients with clinical signs of GCK-MODY, two missense mutations in GCK were found [27].
Recently an approach to detect and evaluate the pathogenic variants using targeted panel sequencing was reported by Park et al. [14]. On genetic testing of 109 suspected monogenic diabetes candidates among 2,090 patients with non-T1DM, using the panel including the exonic and untranslated regions of 30 genes known to cause monogenic diabetes, 14 pathogenic/likely pathogenic variants of common MODY genes were identified. However, 78% of patients did not have a molecular genetic diagnosis, which suggested that the other causes of MODY, such as MODYX may not have been analyzed. The studies on monogenic diabetes in Korean subjects to date are summarized in Table 2.
Table 2.
Studies for monogenic diabetes in Korean subjects
| Gene | Methods | Subjects | Finding | Year | Reference |
|---|---|---|---|---|---|
| HNF1A | SSCP technique | 69 Early onset T2DM | 1/69 (1.5%) Synonymous mutation | 2001 | [18] |
| HNF1A | Sanger sequencing | 16 Early onset T2DM | 1/16 (6.25%) Nonsynonymous mutation (R263L) | 2003 | [19] |
| HNF1A | DNA chip | 22 Early onset T2DM | 1/22 (4.5%) Promoter polymorphism, non-segregating | 2004 | [20] |
| HNF4A, GCK, HNF1A | Sanger sequencing | 23 MODY, 17 early onset T2DM | 2/40 (5%) HNF1A (P393fsdelC, promoter) | 2006 | [21] |
| 1/40 (2.5%) GCK (R191W) | |||||
| 1/40 (2.5%) HNF4A (T130I, polymorphism) | |||||
| HNF1A | DNA chip | 25 Early onset T2DM | 1/25 (4%) Promoter polymorphism | 2008 | [22] |
| HNF1A | Sanger sequencing | 96 GDM | 5/96 (5.2%) 2 Promoter, Arg278Gln, Pro300pro, IVS5 +106A>G | 2008 | [23] |
| HNF1B | Sanger sequencing | 1 MODY | P159L mutation | 2014 | [24] |
| PTPRD, SYT9, WFS1 | WES | 6 MODY | 3 Variants (Thr207Ile in PTPRD, Gln187Glu in SYT9, Val509Gly in WFS1) | 2015 | [25] |
| HNF4A, ABCC8, HNF1A | WES | 28 Early onset T2DM | 4 Pathogenic/likely pathogenic variants (Leu319Pro in HNF4A, His103Tyr and Arg74Gln in ABCC8, Leu139Val in HNF1A) | 2016 | [26] |
| 6 Non-silent variants | |||||
| GCK | WES | 3 Suspected MODY | 2 Variants (Leu30Pro, Ser83Leu) | 2017 | [27] |
| GCK, HNF1A, HNF4A, HNF1B | Targeted panel sequencing | 109 Suspected monogenic diabetes | 14/109 (12.8%): MODY (7 GCK, 3 HNF1A, 3 HNF4A, 1 HNF1B) | 2019 | [14] |
| 5/109 (4.6%): mitochondrial MT-TL1 | |||||
| 4/109 (3.7%): WFS1, INS, ABCC8, FOXP3 |
![]()
Go to :
CLINICAL CHARACTERISTICS AND TREATMENTS ACCORDING TO MODY SUBTYPES
The summary of the clinical characteristics and treatment of MODY to date is described in Table 1. The pathophysiology and clinical characteristics of the most common types of MODY: GCK-MODY, HNF1A-MODY, and HNF4A-MODY—are described below. More specific information about each MODY subtype, including the more rare ones, is available in recent review articles [1,28].
GCK-MODY
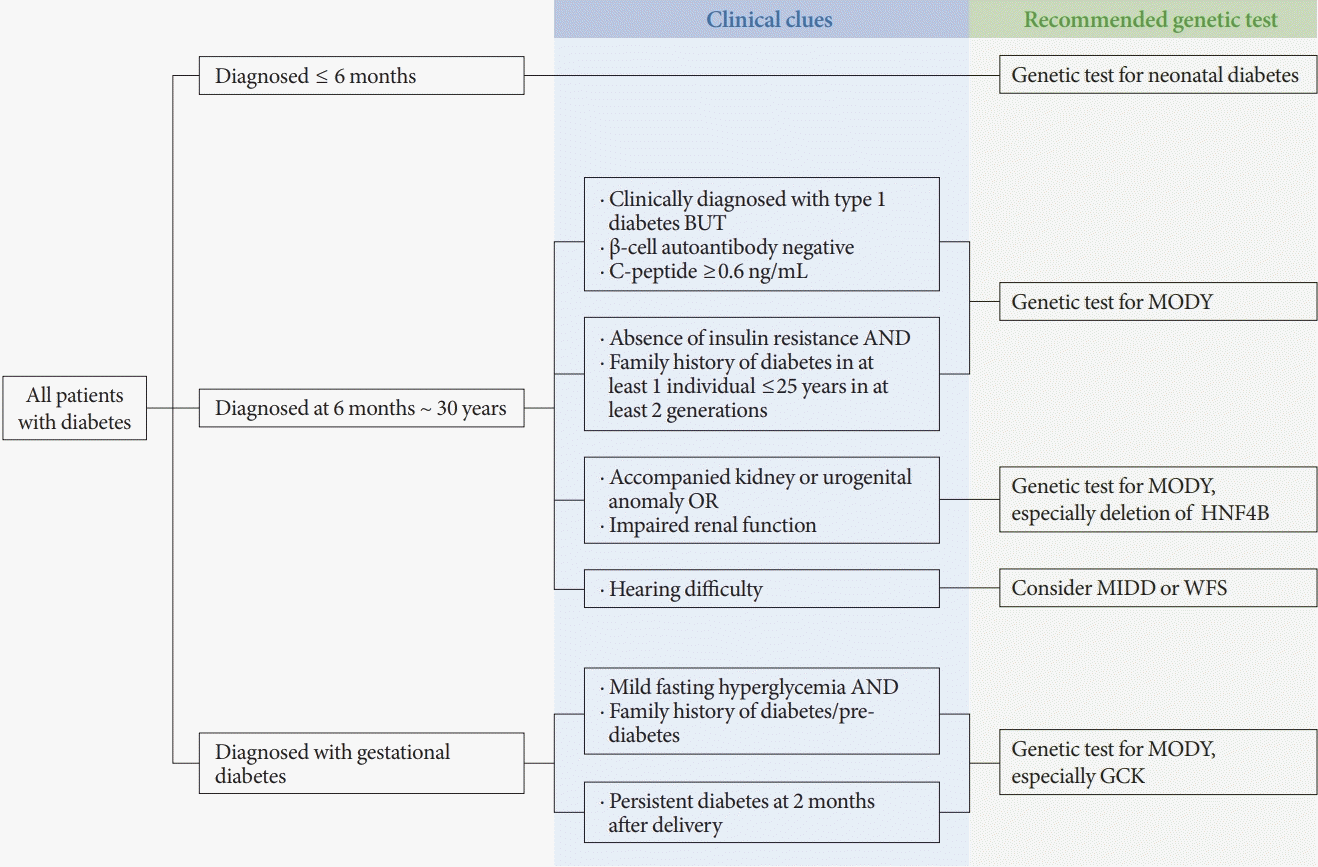
GCK, the β-cell glucose sensor, is a key enzyme that catalyzes glucose to glucose-6-phosphate and controls glucose-mediated insulin secretion. Heterozygous inactivating mutations in GCK gene cause GCK-MODY. More than 600 mutations in more than 1,000 families have been identified and these lead to both hypoglycemia and hyperglycemia [29]. People with GCK-MODY show mild fasting hyperglycemia (5.6 to 8.0 mmol/L, HbA1c range 5.6% to 7.3%) [30], because they have a problem with glucose sensing and their glucose homeostasis is set at a higher range. Patients are usually asymptomatic, so the majority are discovered by routine examinations during pregnancy or school-based urine glucose screening tests. In very rare conditions, a complete deficiency of GCK is reported as permanent neonatal diabetes [31]. In GCK-MODY, microvascular complications [32] are rare and macrovascular complications [33] are similar to those in the general population. Treatment is not recommended except in the case of pregnancy, because glucose-lowering therapy is ineffective [34] and there is a lack of long-term complications. However, it should be noted that T2DM can coexist with GCK-MODY and may be a major risk factor for further morbidities [35]. In pregnancy, fetal growth is determined by whether the mutation is inherited [36]. If the fetus does not inherit the mutation, it will sense the maternal glucose as high and increase insulin secretion which results in increased birth weight. Thus, insulin treatment of the mother is required when fetal overgrowth is detected on fetal growth scans. Since the management of hyperglycemia during pregnancy and after delivery could be altered according to the diagnosis of GCK-MODY, genetic testing should be considered when the GCK mutation is suspected in a woman with GDM (Fig. 1).
HNF1A-MODY
HNF1A, a homeodomain-containing transcription factor, is expressed in the pancreatic β-cells, kidney, liver, and intestine. HNF1A knockout mice develop diabetes because of impaired β-cell response to glucose [37], and also have an abnormal architecture of pancreatic islet [38]. HNF1A is a polymorphic gene without any specified mutation hot-spot [39], and more than 400 different variants have been identified [40]. HNF1A mutations demonstrate high penetrance; 63% of carriers develop diabetes by the age of 25, and almost all of the carriers develop diabetes by the age of 55 [41]. Hyperglycemia induced by heterozygous HNF1A mutations might be severe and progressive, and the risk of long-term micro and macrovascular complications is similar to that of T1DM and T2DM [42]. As patients with HNF1A-MODY show marked sensitivity to sulfonylurea [43-45], low-dose sulfonylurea is recommended as the first-line treatment. In an observational study, 79% of patients could be switched to sulfonylurea from insulin therapy after the correct genetic diagnosis of MODY [46]. Although some patients eventually took insulin treatment, the majority of patients maintained good glycemic control for many years with sulfonylurea [46].
HNF4A-MODY
HNF4A is a transcription factor that is expressed in the liver, intestine, kidney, and pancreatic islets. It regulates genes that are essential to glucose transport and metabolism [47]. More than 100 mutations of HNF4A have been identified so far [40]. The clinical nature of progressive β-cell dysfunction and response to sulfonylureas in heterozygous HNF4A mutations are similar to HNF1A-MODY [48]. Heterozygous HNF4A mutations are associated with significant fetal macrosomia due to increased insulin secretion in utero and the hyperinsulinemia results in neonatal hypoglycemia and diabetes later in life [49]. Therefore, close monitoring of the offspring of an affected mother is recommended in patients with HNF4A-MODY.
Go to :
DIAGNOSTIC APPROACHES OF MONOGENIC DIABETES
Candidates for genetic testing: who should have genetic testing for monogenic diabetes?
As NGS-based targeted panel sequencing is readily available, genetic screening is expected to be performed in more cases. However, additional costs for genetic testing are present, and interpretation of the sequence results remains an issue. The clinical criteria for genetic testing for MODY are not well established [50,51], and there are no concrete cutoff values for the age of diagnosis and BMI. Therefore, clinicians should consider who would be the best candidate for genetic testing for MODY.
Guidelines [52] were established in 2008 by European clinicians and scientists for clinical suspicion of diabetes caused by mutations in the GCK, HNF1A, and HNF4A genes, which are the most common forms of MODY. The clinician’s suspicion for MODY should arise from the atypical features of T1DM and T2DM in childhood, adolescence, or young adulthood [52,53]. Features atypical for T1DM are as follows [54]: absence of pancreatic islet autoantibodies [55], measurable C-peptide in the presence of hyperglycemia [56,57], low insulin requirement for treatment and lack of diabetic ketoacidosis. Features atypical for T2DM include lack of insulin resistance and metabolic syndrome [58,59], young-onset before <45 years, normal weight or mildly overweight, and less frequent insulin use [59]. There are at least five generally accepted diagnostic criteria for MODY [60]: (1) age at diagnosis <25 years in at least one family member; (2) autosomal dominant inheritance across three generations; (3) absence of insulin therapy within 5 years of diagnosis; (4) insulin level within the normal range (plasma insulin ≥2.0 μIU/mL or plasma C-peptide ≥0.6 ng/mL); and (5) not obese (BMI <25 kg/m2).
One suggestive modality for selecting the candidate for the genetic test is the MODY probability calculator [61] which is available online (https://www.diabetesgenes.org/mody-probability-calculator/). It uses eight pieces of clinical information and shows improved sensitivity (91% vs. 72%) and specificity (94% vs. 91%) for detecting MODY compared to standard clinical criteria including diagnosis of patients under 25 years and family history [61]. Although the prediction models were derived from the logistic regression of data on Europeans mostly, it validated that Korean MODY patients showed a significantly high probability [14] as shown in UK patients [62].
Fig. 1 shows an algorithm that recommends genetic testing based on clinical clues suggesting monogenic diabetes. If the onset of diabetes is within the first 6 months of life, genetic testing for neonatal diabetes should be conducted because T1DM usually occurs after then. Genetic tests for MODY should be conducted when a clinician encounters T1DM or T2DM patients with atypical features as mentioned above: (1) the absence of pancreatic autoantibodies and measurable C-peptide levels in T1DM, and (2) the absence of insulin resistance and family history of diabetes in at least one individual aged ≤25 years in at least two generations. When a urogenital anomaly is accompanied in a situation where MODY is suspected, it may be helpful to conduct targeted testing for the HNF4B mutation [63]. In addition, there are some specific clinical features for other rare monogenic diabetes. In the case of MIDD, a rare mitochondrial disorder caused by a genetic mutation in transfer RNA, patients have both a defect in insulin secretion and sensorineural hearing loss [64]. Another syndromic disorder, WFS comes with diabetes insipidus, diabetes mellitus, optic atrophy, and sensorineural deafness [65]. When a subject has hearing difficulty, MIDD or WFS could be the cause. Patients with GCK-MODY show mild fasting hyperglycemia and are usually asymptomatic. As the majority of patients are discovered on routine examinations during pregnancy, woman with diagnosed GDM who have family history of diabetes or persistent diabetes after delivery could have GCK-MODY.
How to interpret the genetic test result
Depending on the target region size, sequencing usually identifies a large number of coding variants in the gene of interest. It is crucial to discern whether the identified variant is the true pathogenic variant or just a bystander. One of the good starting points would be to lookup the variant in the ClinVar database (https://www.ncbi.nlm.nih.gov/clinvar/). This is a public database that archives the relationship between a specific genetic variant and disease status. The pathogenicity of the variant and its evidence are submitted by the provider and reviewed by expert panels. If the identified variant is novel, it should be evaluated for its pathogenicity. Recently the American College of Medical Genetics and Genomics and the Association for Molecular Pathology (ACMG-AMP) [66] has published guidelines for the interpretation of sequence variants to improve the efficacy of clinical genetics. The ACMG-AMP guideline classifies variants as pathogenic, likely pathogenic, variant with uncertain significance, likely benign, and benign. The evidence attributes are derived from population allele frequency, computational prediction, functional studies, segregation analysis, allelic data, etc. Population allele frequency serves as an important criterion to filter out pathogenic variants. This information could be obtained from a genome aggregation database (gnomAD, https://gnomad.broadinstitute.org/), which also includes the allele frequency in Koreans. However, it would be important to have a larger database for Korean specific allele frequency and also to archive variants that are associated with monogenic diabetes.
Non-genetic biomarkers for MODY
There have been many efforts to discover non-genetic biomarkers to improve the diagnosis rate of MODY with efficient and cheap methods [67]. The approach using biomarkers is expected to provide additional clinical suspicions of MODY and narrow down candidates for molecular testing. Among the suggested biomarkers so far, high-sensitivity C-reactive protein (hs-CRP) has emerged as a novel biomarker after the association between the alteration in hs-CRP levels and common variants in HNF1A was reported with genome-wide association studies (GWAS) [68,69]. The level of hs-CRP was significantly lower in HNF1A-MODY patients than in any other group—T1DM, T2DM, GCK-MODY, and normal glucose-tolerant participants—in several studies of the European population [70-73]. Because hs-CRP has been widely used in many clinical settings, it would be a cost-effective screening test to discriminate HNF1A-MODY from other types of diabetes when combined with current clinical diagnosis. However, the cutoff values for differential diagnosis are not well established [70,72]. Additionally, there are controversial results regarding Asian ethnicities because of various factors, such as BMI and race influence on the level of hs-CRP. In a Japanese HNF1A-MODY patient, the serum hs-CRP level was lower than that observed in T1DM and T2DM [74]. A study in Singapore which mostly involved Chinese and Indian races did not show improvement in the diagnostic yield with the hs-CRP criteria (1/37, 2.7%) when compared with the traditional clinical criteria (5/125, 4%) [75]. Thus, the efficacy of hs-CRP as a diagnostic criterion and the appropriate cutoff values for clinical feasibility, especially in the Asian population need to be elucidated.
In addition to hs-CRP, plasma fucosylated glycans is a promising candidate for an HNF1A diagnostic marker [73,76]. A study combining GWAS and glycomics analysis revealed that HNF1A regulates multiple steps in fucosylation [77] which affects N-glycan levels in humans. The discriminative performance of both total plasma N-glycome and some glycan groups showed higher power with area under the curves, at least more than 0.87, in distinguishing patients with damaging HNF1A alleles from those without risk alleles [73]. Apolipoprotein M (apoM), a lipoprotein that is associated with the high-density lipoprotein particle is another protein encoded by a gene for which HNF1A acts as a transcriptional activator. Serum apoM level was lower in the HNF1A-MODY group compared with the control participants [78] and the T1DM group [79]. However, a subsequent study could not replicate the result [80], and only women with HNF1A mutations had 10% lower serum apoM levels compared with controls in another study [81]. Due to a variety of techniques and subject identifications used for analysis, discrepancies could be observed between studies using biomarkers. Further work and standard techniques are needed to investigate the role of apoM as a useful biomarker for HNF1A-MODY.
Recently microRNAs (miRNAs) have emerged as potential regulators of gene functions associated with diabetes [82-84]; therefore, approaches using miRNAs as biomarkers for differentiating MODY from other types of diabetes have been used in vivo [85,86] and in vitro [87]. Although the miRNAs should be verified in large population studies in order to be used as biomarkers in a clinical setting, if miRNAs are involved in the pathophysiology of MODY and its antagomirs can be used to correct pathognomonic pathways, they can be used as therapeutic targets for MODY.
Go to :
CONCLUSION
Monogenic diabetes in Asians is estimated to represent around 1 to 6% of all diabetes cases. In Korea, there have been 11 studies to date which focused on identifying monogenic diabetes. In the recent largest study in 2019 using targeted exome panel sequencing, a molecular diagnosis rate was 21.1% for monogenic diabetes in clinically suspected patients. As the study design influences the prevalence and genetic diagnosis rate, the definite prevalence and difference due to ethnicity could not be concluded. For further established results, it is necessary to conduct a pan-Asian registration and a joint research to accumulate MODY frequency and allele frequency data for Asians. The most important step in the diagnosis of monogenic diabetes is to identify candidates for the genetic test. The physician should be familiar with the clinical clues of monogenic diabetes and have a clear understanding of fundamental principles for interpreting the results. The suggestive algorithm and non-genetic tools could help the diagnostic approach.
Go to :
 XML Download
XML Download