1. Younossi ZM, Koenig AB, Abdelatif D, Fazel Y, Henry L, Wymer M. Global epidemiology of nonalcoholic fatty liver disease: meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology. 2016; 64:73–84.

2. Angulo P, Kleiner DE, Dam-Larsen S, Adams LA, Bjornsson ES, Charatcharoenwitthaya P, Mills PR, Keach JC, Lafferty HD, Stahler A, Haflidadottir S, Bendtsen F. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology. 2015; 149:389–97.
3. Musso G, Gambino R, Cassader M, Pagano G. Meta-analysis: natural history of non-alcoholic fatty liver disease (NAFLD) and diagnostic accuracy of non-invasive tests for liver disease severity. Ann Med. 2011; 43:617–49.
4. Vernon G, Baranova A, Younossi ZM. Systematic review: the epidemiology and natural history of non-alcoholic fatty liver disease and non-alcoholic steatohepatitis in adults. Aliment Pharmacol Ther. 2011; 34:274–85.
5. Rafiq N, Bai C, Fang Y, Srishord M, McCullough A, Gramlich T, Younossi ZM. Long-term follow-up of patients with nonalcoholic fatty liver. Clin Gastroenterol Hepatol. 2009; 7:234–8.
6. Sayiner M, Koenig A, Henry L, Younossi ZM. Epidemiology of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis in the united states and the rest of the world. Clin Liver Dis. 2016; 20:205–14.
7. GBD 2015 Mortality and Causes of Death Collaborators. Global, regional, and national life expectancy, all-cause mortality, and cause-specific mortality for 249 causes of death, 1980-2015: a systematic analysis for the Global Burden of Disease Study 2015. Lancet. 2016; 388:1459–544.
8. Younossi ZM, Stepanova M, Rafiq N, Makhlouf H, Younoszai Z, Agrawal R, Goodman Z. Pathologic criteria for nonalcoholic steatohepatitis: interprotocol agreement and ability to predict liver-related mortality. Hepatology. 2011; 53:1874–82.
9. Hossain N, Stepanova M, Afendy A, Nader F, Younossi Y, Rafiq N, Goodman Z, Younossi ZM. Non-alcoholic steatohepatitis (NASH) in patients with polycystic ovarian syndrome (PCOS). Scand J Gastroenterol. 2011; 46:479–84.
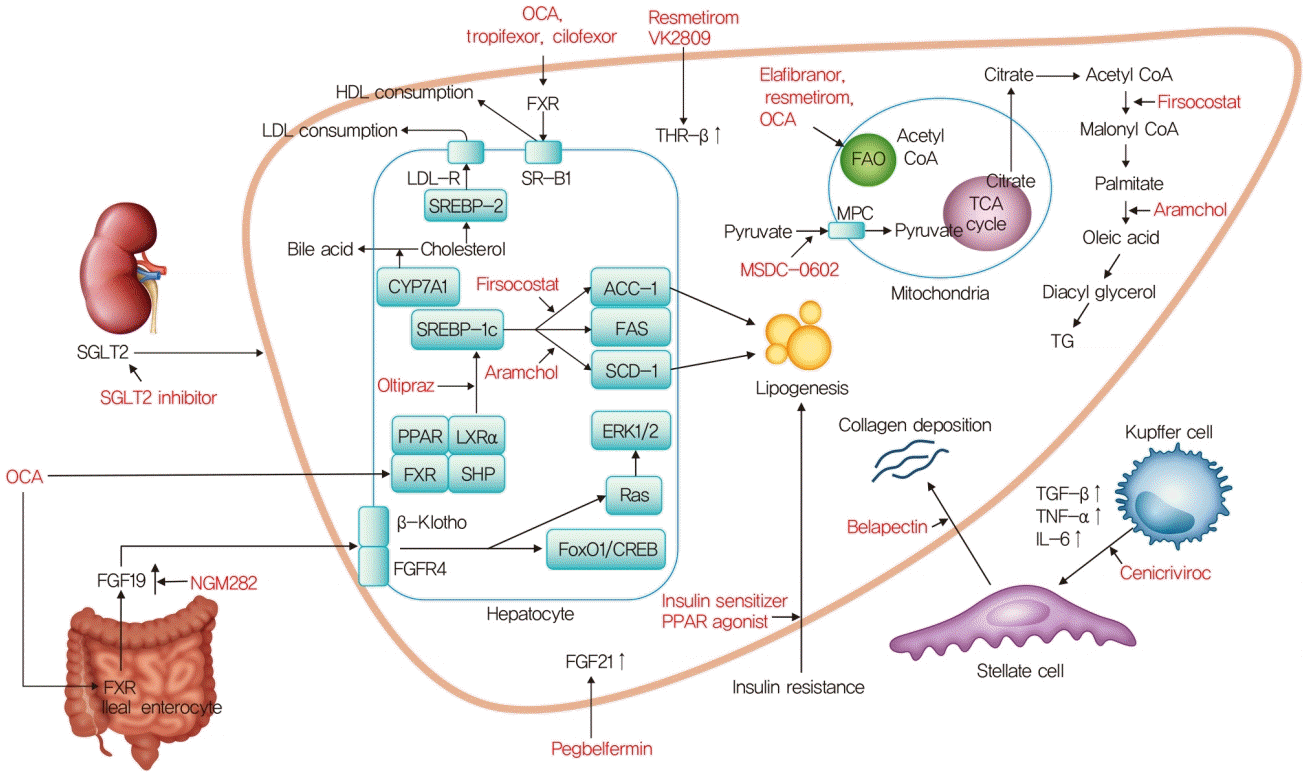
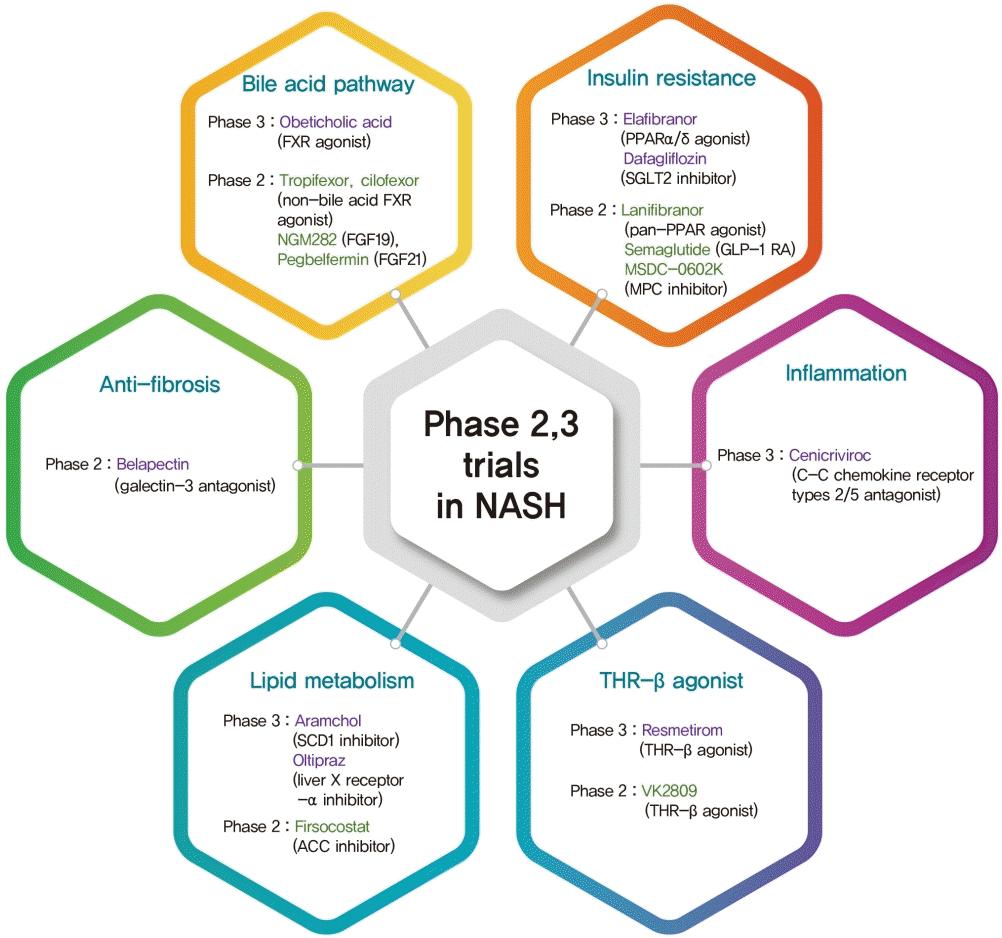
10. Konerman MA, Jones JC, Harrison SA. Pharmacotherapy for NASH: current and emerging. J Hepatol. 2018; 68:362–75.
11. Yoo JJ, Kim W, Kim MY, Jun DW, Kim SG, Yeon JE, Lee JW, Cho YK, Park SH, Sohn JH JH; the Korean Association for the Study of the Liver (KASL)-Korea Nonalcoholic fatty liver Study Group (KNSG). Recent research trends and updates on nonalcoholic fatty liver disease. Clin Mol Hepatol. 2019; 25:1–11.
12. Cusi K, Orsak B, Bril F, Lomonaco R, Hecht J, Ortiz-Lopez C, Tio F, Hardies J, Darland C, Musi N, Webb A, Portillo-Sanchez P. Long-term pioglitazone treatment for patients with nonalcoholic steatohepatitis and prediabetes or type 2 diabetes mellitus: a randomized trial. Ann Intern Med. 2016; 165:305–15.
13. Aithal GP, Thomas JA, Kaye PV, Lawson A, Ryder SD, Spendlove I, Austin AS, Freeman JG, Morgan L, Webber J. Randomized, placebo-controlled trial of pioglitazone in nondiabetic subjects with nonalcoholic steatohepatitis. Gastroenterology. 2008; 135:1176–84.
14. Sanyal AJ, Chalasani N, Kowdley KV, McCullough A, Diehl AM, Bass NM, Neuschwander-Tetri BA, Lavine JE, Tonascia J, Unalp A, Van Natta M, Clark J, Brunt EM, Kleiner DE, Hoofnagle JH, Robuck PR; NASH CRN. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N Engl J Med. 2010; 362:1675–85.
15. Armstrong MJ, Gaunt P, Aithal GP, Barton D, Hull D, Parker R, Hazlehurst JM, Guo K; LEAN trial team, Abouda G, Aldersley MA, Stocken D, Gough SC, Tomlinson JW, Brown RM, Hubscher SG, Newsome PN. Liraglutide safety and efficacy in patients with non-alcoholic steatohepatitis (LEAN): a multicentre, double-blind, randomised, placebo-controlled phase 2 study. Lancet. 2016; 387:679–90.
16. Caldwell S. NASH therapy: omega 3 supplementation, vitamin E, insulin sensitizers and statin drugs. Clin Mol Hepatol. 2017; 23:103–8.
17. Bjelakovic G, Nikolova D, Gluud LL, Simonetti RG, Gluud C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: systematic review and meta-analysis. JAMA. 2007; 297:842–57.
18. Abner EL, Schmitt FA, Mendiondo MS, Marcum JL, Kryscio RJ. Vitamin E and all-cause mortality: a meta-analysis. Curr Aging Sci. 2011; 4:158–70.
19. Schurks M, Glynn RJ, Rist PM, Tzourio C, Kurth T. Effects of vitamin E on stroke subtypes: meta-analysis of randomised controlled trials. BMJ. 2010; 341:c5702.
20. Klein EA, Thompson IM Jr, Tangen CM, Crowley JJ, Lucia MS, Goodman PJ, Minasian LM, Ford LG, Parnes HL, Gaziano JM, Karp DD, Lieber MM, Walther PJ, Klotz L, Parsons JK, Chin JL, Darke AK, Lippman SM, Goodman GE, Meyskens FL Jr, Baker LH. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011; 306:1549–56.
21. Vilar-Gomez E, Martinez-Perez Y, Calzadilla-Bertot L, Torres-Gonzalez A, Gra-Oramas B, Gonzalez-Fabian L, Friedman SL, Diago M, Romero-Gomez M. Weight loss through lifestyle modification significantly reduces features of nonalcoholic steatohepatitis. Gastroenterology. 2015; 149:367–78.
22. St George A, Bauman A, Johnston A, Farrell G, Chey T, George J. Independent effects of physical activity in patients with nonalcoholic fatty liver disease. Hepatology. 2009; 50:68–76.
23. Sung KC, Ryu S, Lee JY, Kim JY, Wild SH, Byrne CD. Effect of exercise on the development of new fatty liver and the resolution of existing fatty liver. J Hepatol. 2016; 65:791–7.
24. Chalasani N, Younossi Z, Lavine JE, Charlton M, Cusi K, Rinella M, Harrison SA, Brunt EM, Sanyal AJ. The diagnosis and management of nonalcoholic fatty liver disease: practice guidance from the American Association for the Study of Liver Diseases. Hepatology. 2018; 67:328–57.
25. Neuschwander-Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, Chalasani N, Dasarathy S, Diehl AM, Hameed B, Kowdley KV, McCullough A, Terrault N, Clark JM, Tonascia J, Brunt EM, Kleiner DE, Doo E; NASH Clinical Research Network. Farnesoid X nuclear receptor ligand obeticholic acid for non-cirrhotic, non-alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo-controlled trial. Lancet. 2015; 385:956–65.
26. Ratziu V, Harrison SA, Francque S, Bedossa P, Lehert P, Serfaty L, Romero-Gomez M, Boursier J, Abdelmalek M, Caldwell S, Drenth J, Anstee QM, Hum D, Hanf R, Roudot A, Megnien S, Staels B, Sanyal A; GOLDEN-505 Investigator Study Group. Elafibranor, an agonist of the peroxisome proliferator-activated receptor-α and -δ, induces resolution of nonalcoholic steatohepatitis without fibrosis worsening. Gastroenterology. 2016; 150:1147–59.
27. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, Francque S, Farrell G, Kowdley KV, Craxi A, Simon K, Fischer L, Melchor-Khan L, Vest J, Wiens BL, Vig P, Seyedkazemi S, Goodman Z, Wong VW, Loomba R, Tacke F, Sanyal A, Lefebvre E. A randomized, placebo-controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology. 2018; 67:1754–67.
28. Harrison SA, Bashir MR, Guy CD, Zhou R, Moylan CA, Frias JP, Alkhouri N, Bansal MB, Baum S, Neuschwander-Tetri BA, Taub R, Moussa SE. Resmetirom (MGL-3196) for the treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2019; 394:2012–24.
29. Ratziu V, Ladron-De-Guevara L, Safadi R, Poordad F, Fuster F, Harrison SA, Arrese M, Fargion S, Ben-Bashat D, Lackner C, Gorfine T, Oren R, Loomba R, Sanyal AJ; the ARREST investigator study group. One-year results of the global phase 2b randomized placebo-controlled ARREST trial of aramchol, a stearoyl CoA desaturase inhibitor, in patients with NASH. Hepatology. 2018; 68:1447A–8A.
30. Pellicciari R, Costantino G, Camaioni E, Sadeghpour BM, Entrena A, Willson TM, Fiorucci S, Clerici C, Gioiello A. Bile acid derivatives as ligands of the farnesoid X receptor: synthesis, evaluation, and structure-activity relationship of a series of body and side chain modified analogues of chenodeoxycholic acid. J Med Chem. 2004; 47:4559–69.
31. Jhaveri MA, Kowdley KV. New developments in the treatment of primary biliary cholangitis: role of obeticholic acid. Ther Clin Risk Manag. 2017; 13:1053–60.
32. Adorini L, Pruzanski M, Shapiro D. Farnesoid X receptor targeting to treat nonalcoholic steatohepatitis. Drug Discov Today. 2012; 17:988–97.
33. Pellicciari R, Fiorucci S, Camaioni E, Clerici C, Costantino G, Maloney PR, Morelli A, Parks DJ, Willson TM. 6Alpha-ethyl-chenodeoxycholic acid (6-ECDCA), a potent and selective FXR agonist endowed with anticholestatic activity. J Med Chem. 2002; 45:3569–72.
34. Mudaliar S, Henry RR, Sanyal AJ, Morrow L, Marschall HU, Kipnes M, Adorini L, Sciacca CI, Clopton P, Castelloe E, Dillon P, Pruzanski M, Shapiro D. Efficacy and safety of the farnesoid X receptor agonist obeticholic acid in patients with type 2 diabetes and nonalcoholic fatty liver disease. Gastroenterology. 2013; 145:574–82.
35. Younossi ZM, Ratziu V, Loomba R, Rinella M, Anstee QM, Goodman Z, Bedossa P, Geier A, Beckebaum S, Newsome PN, Sheridan D, Sheikh MY, Trotter J, Knapple W, Lawitz E, Abdelmalek MF, Kowdley KV, Montano-Loza AJ, Boursier J, Mathurin P, Bugianesi E, Mazzella G, Olveira A, Cortez-Pinto H, Graupera I, Orr D, Gluud LL, Dufour JF, Shapiro D, Campagna J, Zaru L, MacConell L, Shringarpure R, Harrison S, Sanyal AJ; REGENERATE Study Investigators. Obeticholic acid for the treatment of non-alcoholic steatohepatitis: interim analysis from a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2019; 394:2184–96.
36. Pockros PJ, Fuchs M, Freilich B, Schiff E, Kohli A, Lawitz EJ, Hellstern PA, Owens-Grillo J, Van Biene C, Shringarpure R, MacConell L, Shapiro D, Cohen DE. CONTROL: a randomized phase 2 study of obeticholic acid and atorvastatin on lipoproteins in nonalcoholic steatohepatitis patients. Liver Int. 2019; 39:2082–93.
37. Eaton JE, Vuppalanchi R, Reddy R, Sathapathy S, Ali B, Kamath PS. Liver injury in patients with cholestatic liver disease treated with obeticholic acid. Hepatology. 2020; 71:1511–4.
38. Tully DC, Rucker PV, Chianelli D, Williams J, Vidal A, Alper PB, Mutnick D, Bursulaya B, Schmeits J, Wu X, Bao D, Zoll J, Kim Y, Groessl T, McNamara P, Seidel HM, Molteni V, Liu B, Phimister A, Joseph SB, Laffitte B. Discovery of tropifexor (LJN452), a highly potent non-bile acid fxr agonist for the treatment of cholestatic liver diseases and nonalcoholic steatohepatitis (NASH). J Med Chem. 2017; 60:9960–73.
39. Sanyal AJ, Lopez PM, Lawitz E, Kim W, Huang JF, Andreone P, Goh BBG, Chen YC, Ratziu V, Kim YJ, Ryan M, Weltman M, Geier A, Loeffler J, Schaefer FA, Vaidyanathan S, Brass CA. Tropifexor (TXR), an FXR agonist for the treatment of NASH: interim results from first two parts of phase 2b study flight-FXR. Hepatology. 2018; 68:1460A–1A.
40. Sanyal A, Lopez P, Lawitz E, Kim W, Huang JF, Andreone P, Goh GBB, Chen YC, Ratziu V, Kim YJ, Ryan M, Weltman M, Geier A, Loeffler J, Schaefer F, Vaidyanathan S, Martic M, Brass C. SAT-357. Tropifexor, a farnesoid X receptor agonist for the treatment of non-alcoholic steatohepatitis: Interim results based on baseline body mass index from first two parts of Phase 2b study FLIGHT-FXR7. J Hepatol. 2019; 70(1 Suppl):E796–7.
41. Trauner M, Gulamhusein A, Hameed B, Caldwell S, Shiffman ML, Landis C, Eksteen B, Agarwal K, Muir A, Rushbrook S, Lu X, Xu J, Chuang JC, Billin AN, Li G, Chung C, Subramanian GM, Myers RP, Bowlus CL, Kowdley KV. The nonsteroidal farnesoid X receptor agonist cilofexor (GS-9674) improves markers of cholestasis and liver injury in patients with primary sclerosing cholangitis. Hepatology. 2019; 70:788–801.
42. Kirby B, Djedjos CS, Birkebak J, Song Q, Grycz K, Weston J, Subramanian M, Watkins W, Myers RP, Mathias A. Evaluation of the safety and pharmacokinetic effects of the oral, non-steroidal farnesoid X receptor agonist GS-9674 in healthy volunteers. Hepatology. 2016; 63:574A–5A.
43. Lawitz E, Herring R Jr, Younes ZH, Gane E, Ruane P, Schall RA, Jia C, Xu R, Mccolgan B, Djedjos S, Subramanian M, Mchutchison JG, Myers R, Middleton M, Li K, Hellerstein M, Kwo P, Noureddin M, Harrison S. Proof of concept study of an apoptosis-signal regulating kinase (ASK1) inhibitor (selonsertib) in combination with an acetyl-CoA carboxylase inhibitor (GS-0976) or a farnesoid X receptor agonist (GS-9674) in NASH. J Hepatol. 2018; 68 Suppl 1:S57.
44. Patel K, Harrison SA, Elkhashab M, Trotter JF, Herring R, Rojter SE, Kayali Z, Wong VW, Greenbloom S, Jayakumar S, Shiffman ML, Freilich B, Lawitz EJ, Gane EJ, Harting E, Xu J, Billin AN, Chung C, Djedjos CS, Subramanian GM, Myers RP, Middleton MS, Rinella M, Noureddin M. Cilofexor, a nonsteroidal FXR agonist, in patients with noncirrhotic NASH: a phase 2 randomized controlled trial. Hepatology. 2020; 72:58–71.
45. Chianelli D, Rucker PV, Roland J, Tully DC, Nelson J, Liu X, Bursulaya B, Hernandez ED, Wu J, Prashad M, Schlama T, Liu Y, Chu A, Schmeits J, Huang DJ, Hill R, Bao D, Zoll J, Kim Y, Groessl T, McNamara P, Liu B, Richmond W, Sancho-Martinez I, Phimister A, Seidel HM, Badman MK, Joseph SB, Laffitte B, Molteni V. Nidufexor (LMB763), a novel FXR modulator for the treatment of nonalcoholic steatohepatitis. J Med Chem. 2020; 63:3868–80.
46. Erstad DJ, Farrar CT, Ghoshal S, Masia R, Ferreira DS, Chen YI, Choi JK, Wei L, Waghorn PA, Rotile NJ, Tu C, Graham-O’Regan KA, Sojoodi M, Li S, Li Y, Wang G, Corey KE, Or YS, Jiang L, Tanabe KK, Caravan P, Fuchs BC. Molecular magnetic resonance imaging accurately measures the antifibrotic effect of EDP-305, a novel farnesoid X receptor agonist. Hepatol Commun. 2018; 2:821–35.
47. Ornitz DM, Itoh N. The fibroblast growth factor signaling pathway. Wiley Interdiscip Rev Dev Biol. 2015; 4:215–66.
48. Schaap FG, Trauner M, Jansen PL. Bile acid receptors as targets for drug development. Nat Rev Gastroenterol Hepatol. 2014; 11:55–67.
49. Harrison SA, Rinella ME, Abdelmalek MF, Trotter JF, Paredes AH, Arnold HL, Kugelmas M, Bashir MR, Jaros MJ, Ling L, Rossi SJ, DePaoli AM, Loomba R. NGM282 for treatment of non-alcoholic steatohepatitis: a multicentre, randomised, double-blind, placebo-controlled, phase 2 trial. Lancet. 2018; 391:1174–85.
50. Harrison SA, Rossi SJ, Paredes AH, Trotter JF, Bashir MR, Guy CD, Banerjee R, Jaros MJ, Owers S, Baxter BA, Ling L, DePaoli AM. NGM282 improves liver fibrosis and histology in 12 weeks in patients with nonalcoholic steatohepatitis. Hepatology. 2020; 71:1198–212.
51. Cyphert HA, Ge X, Kohan AB, Salati LM, Zhang Y, Hillgartner FB. Activation of the farnesoid X receptor induces hepatic expression and secretion of fibroblast growth factor 21. J Biol Chem. 2012; 287:25123–38.
52. Kharitonenkov A, Larsen P. FGF21 reloaded: challenges of a rapidly growing field. Trends Endocrinol Metab. 2011; 22:81–6.
53. Sonoda J, Chen MZ, Baruch A. FGF21-receptor agonists: an emerging therapeutic class for obesity-related diseases. Horm Mol Biol Clin Investig. 2017; 30:20170002.
54. Sanyal A, Charles ED, Neuschwander-Tetri BA, Loomba R, Harrison SA, Abdelmalek MF, Lawitz EJ, Halegoua-DeMarzio D, Kundu S, Noviello S, Luo Y, Christian R. Pegbelfermin (BMS-986036), a PEGylated fibroblast growth factor 21 analogue, in patients with non-alcoholic steatohepatitis: a randomised, double-blind, placebo-controlled, phase 2a trial. Lancet. 2019; 392:2705–17.
55. Berger J, Moller DE. The mechanisms of action of PPARs. Annu Rev Med. 2002; 53:409–35.
56. Pawlak M, Lefebvre P, Staels B. Molecular mechanism of PPARα action and its impact on lipid metabolism, inflammation and fibrosis in non-alcoholic fatty liver disease. J Hepatol. 2015; 62:720–33.
57. Francque S, Verrijken A, Caron S, Prawitt J, Paumelle R, Derudas B, Lefebvre P, Taskinen MR, Van Hul W, Mertens I, Hubens G, Van Marck E, Michielsen P, Van Gaal L, Staels B. PPARα gene expression correlates with severity and histological treatment response in patients with non-alcoholic steatohepatitis. J Hepatol. 2015; 63:164–73.
58. Bojic LA, Huff MW. Peroxisome proliferator-activated receptor δ: a multifaceted metabolic player. Curr Opin Lipidol. 2013; 24:171–7.
59. Riserus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, Fang Z, Hegde P, Richards D, Sarov-Blat L, Strum JC, Basu S, Cheeseman J, Fielding BA, Humphreys SM, Danoff T, Moore NR, Murgatroyd P, O’Rahilly S, Sutton P, Willson T, Hassall D, Frayn KN, Karpe F. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008; 57:332–9.
60. Boubia B, Poupardin O, Barth M, Binet J, Peralba P, Mounier L, Jacquier E, Gauthier E, Lepais V, Chatar M, Ferry S, Thourigny A, Guillier F, Llacer J, Amaudrut J, Dodey P, Lacombe O, Masson P, Montalbetti C, Wettstein G, Luccarini JM, Legendre C, Junien JL, Broqua P. Design, synthesis, and evaluation of a novel series of indole sulfonamide peroxisome proliferator activated receptor (PPAR) α/γ/δ triple activators: discovery of lanifibranor, a new antifibrotic clinical candidate. J Med Chem. 2018; 61:2246–65.
61. American Diabetes Association. 8. Pharmacologic approaches to glycemic treatment: standards of medical care in diabetes-2018. Diabetes Care. 2018; 41:S73–85.
62. Eng C, Kramer CK, Zinman B, Retnakaran R. Glucagon-like peptide-1 receptor agonist and basal insulin combination treatment for the management of type 2 diabetes: a systematic review and meta-analysis. Lancet. 2014; 384:2228–34.
63. Eguchi Y, Kitajima Y, Hyogo H, Takahashi H, Kojima M, Ono M, Araki N, Tanaka K, Yamaguchi M, Matsuda Y, Ide Y, Otsuka T, Ozaki I, Ono N, Eguchi T, Anzai K; Japan Study Group for NAFLD (JSG-NAFLD). Pilot study of liraglutide effects in non-alcoholic steatohepatitis and non-alcoholic fatty liver disease with glucose intolerance in Japanese patients (LEAN-J). Hepatol Res. 2015; 45:269–78.
64. Armstrong MJ, Houlihan DD, Rowe IA, Clausen WH, Elbrønd B, Gough SC, Tomlinson JW, Newsome PN. Safety and efficacy of liraglutide in patients with type 2 diabetes and elevated liver enzymes: individual patient data meta-analysis of the LEAD program. Aliment Pharmacol Ther. 2013; 37:234–42.
65. Kenny PR, Brady DE, Torres DM, Ragozzino L, Chalasani N, Harrison SA. Exenatide in the treatment of diabetic patients with non-alcoholic steatohepatitis: a case series. Am J Gastroenterol. 2010; 105:2707–9.
66. Buse JB, Klonoff DC, Nielsen LL, Guan X, Bowlus CL, Holcombe JH, Maggs DG, Wintle ME. Metabolic effects of two years of exenatide treatment on diabetes, obesity, and hepatic biomarkers in patients with type 2 diabetes: an interim analysis of data from the open-label, uncontrolled extension of three double-blind, placebo-controlled trials. Clin Ther. 2007; 29:139–53.
67. Shimizu M, Suzuki K, Kato K, Jojima T, Iijima T, Murohisa T, Iijima M, Takekawa H, Usui I, Hiraishi H, Aso Y. Evaluation of the effects of dapagliflozin, a sodium-glucose co-transporter-2 inhibitor, on hepatic steatosis and fibrosis using transient elastography in patients with type 2 diabetes and non-alcoholic fatty liver disease. Diabetes Obes Metab. 2019; 21:285–92.
68. Kurinami N, Sugiyama S, Yoshida A, Hieshima K, Miyamoto F, Kajiwara K, Jinnouch K, Jinnouchi T, Jinnouchi H. Dapagliflozin significantly reduced liver fat accumulation associated with a decrease in abdominal subcutaneous fat in patients with inadequately controlled type 2 diabetes mellitus. Diabetes Res Clin Pract. 2018; 142:254–63.
69. Soccio RE, Chen ER, Lazar MA. Thiazolidinediones and the promise of insulin sensitization in type 2 diabetes. Cell Metab. 2014; 20:573–91.
70. Chen Z, Vigueira PA, Chambers KT, Hall AM, Mitra MS, Qi N, McDonald WG, Colca JR, Kletzien RF, Finck BN. Insulin resistance and metabolic derangements in obese mice are ameliorated by a novel peroxisome proliferator-activated receptor γ-sparing thiazolidinedione. J Biol Chem. 2012; 287:23537–48.
71. McCommis KS, Hodges WT, Brunt EM, Nalbantoglu I, McDonald WG, Holley C, Fujiwara H, Schaffer JE, Colca JR, Finck BN. Targeting the mitochondrial pyruvate carrier attenuates fibrosis in a mouse model of nonalcoholic steatohepatitis. Hepatology. 2017; 65:1543–56.
72. Colca JR, McDonald WG, Cavey GS, Cole SL, Holewa DD, Brightwell-Conrad AS, Wolfe CL, Wheeler JS, Coulter KR, Kilkuskie PM, Gracheva E, Korshunova Y, Trusgnich M, Karr R, Wiley SE, Divakaruni AS, Murphy AN, Vigueira PA, Finck BN, Kletzien RF. Identification of a mitochondrial target of thiazolidinedione insulin sensitizers (mTOT): relationship to newly identified mitochondrial pyruvate carrier proteins. PLoS One. 2013; 8:e61551.
73. Colca JR, McDonald WG, Adams WJ. MSDC-0602K, a metabolic modulator directed at the core pathology of non-alcoholic steatohepatitis. Expert Opin Investig Drugs. 2018; 27:631–6.
74. Harrison SA, Alkhouri N, Davison BA, Sanyal A, Edwards C, Colca JR, Lee BH, Loomba R, Cusi K, Kolterman O, Cotter G, Dittrich HC. Insulin sensitizer MSDC-0602K in non-alcoholic steatohepatitis: a randomized, double-blind, placebo-controlled phase IIb study. J Hepatol. 2020; 72:613–26.
75. Mossanen JC, Krenkel O, Ergen C, Govaere O, Liepelt A, Puengel T, Heymann F, Kalthoff S, Lefebvre E, Eulberg D, Luedde T, Marx G, Strassburg CP, Roskams T, Trautwein C, Tacke F. Chemokine (C-C motif) receptor 2-positive monocytes aggravate the early phase of acetaminophen-induced acute liver injury. Hepatology. 2016; 64:1667–82.
76. Lefebvre E, Moyle G, Reshef R, Richman LP, Thompson M, Hong F, Chou HL, Hashiguchi T, Plato C, Poulin D, Richards T, Yoneyama H, Jenkins H, Wolfgang G, Friedman SL. Antifibrotic effects of the dual CCR2/CCR5 antagonist cenicriviroc in animal models of liver and kidney fibrosis. PLoS One. 2016; 11:e0158156.
77. Schwabe RF, Bataller R, Brenner DA. Human hepatic stellate cells express CCR5 and RANTES to induce proliferation and migration. Am J Physiol Gastrointest Liver Physiol. 2003; 285:G949–58.
78. Friedman S, Sanyal A, Goodman Z, Lefebvre E, Gottwald M, Fischer L, Ratziu V. Efficacy and safety study of cenicriviroc for the treatment of non-alcoholic steatohepatitis in adult subjects with liver fibrosis: CENTAUR Phase 2b study design. Contemp Clin Trials. 2016; 47:356–65.
79. Sinha RA, Bruinstroop E, Singh BK, Yen PM. Nonalcoholic fatty liver disease and hypercholesterolemia: roles of thyroid hormones, metabolites, and agonists. Thyroid. 2019; 29:1173–91.
80. Bohinc BN, Michelotti G, Xie G, Pang H, Suzuki A, Guy CD, Piercy D, Kruger L, Swiderska-Syn M, Machado M, Pereira T, Zavacki AM, Abdelmalek M, Diehl AM. Repair-related activation of hedgehog signaling in stromal cells promotes intrahepatic hypothyroidism. Endocrinology. 2014; 155:4591–601.
81. Loomba R, Neutel J, Mohseni R, Bernard D, Severance R, Dao M, Saini S, Margaritescu C, Homer K, Tran B, Mancini M, Masamune H, Lian B. VK2809. A novel liver-directed thyroid receptor beta agonist, significantly reduces liver fat with both low and high doses in patients with non-alcoholic fatty liver disease: a phase 2 randomized, placebo-controlled trial. J Hepatol. 2019; 70(1 Suppl):E150–1.
82. Leikin-Frenkel A, Gonen A, Shaish A, Goldiner I, Leikin-Gobbi D, Konikoff FM, Harats D, Gilat T. Fatty acid bile acid conjugate inhibits hepatic stearoyl coenzyme A desaturase and is non-atherogenic. Arch Med Res. 2010; 41:397–404.
83. Abu-Elheiga L, Brinkley WR, Zhong L, Chirala SS, Woldegiorgis G, Wakil SJ. The subcellular localization of acetyl-CoA carboxylase 2. Proc Natl Acad Sci U S A. 2000; 97:1444–9.
84. Brooks SC 3rd, Brooks JS, Lee WH, Lee MG, Kim SG. Therapeutic potential of dithiolethiones for hepatic diseases. Pharmacol Ther. 2009; 124:31–43.
85. Winder WW, Hardie DG. AMP-activated protein kinase, a metabolic master switch: possible roles in type 2 diabetes. Am J Physiol. 1999; 277:E1–10.
86. Kim W, Kim BG, Lee JS, Lee CK, Yeon JE, Chang MS, Kim JH, Kim H, Yi S, Lee J, Cho JY, Kim SG, Lee JH, Kim YJ. Randomised clinical trial: the efficacy and safety of oltipraz, a liver X receptor alpha-inhibitory dithiolethione in patients with non-alcoholic fatty liver disease. Aliment Pharmacol Ther. 2017; 45:1073–83.
87. Abu-Elheiga L, Jayakumar A, Baldini A, Chirala SS, Wakil SJ. Human acetyl-CoA carboxylase: characterization, molecular cloning, and evidence for two isoforms. Proc Natl Acad Sci U S A. 1995; 92:4011–5.
88. Harriman G, Greenwood J, Bhat S, Huang X, Wang R, Paul D, Tong L, Saha AK, Westlin WF, Kapeller R, Harwood HJ Jr. Acetyl-CoA carboxylase inhibition by ND-630 reduces hepatic steatosis, improves insulin sensitivity, and modulates dyslipidemia in rats. Proc Natl Acad Sci U S A. 2016; 113:E1796–805.
89. Harwood HJ Jr. Acetyl-CoA carboxylase inhibition for the treatment of metabolic syndrome. Curr Opin Investig Drugs. 2004; 5:283–9.
90. Harwood HJ Jr. Treating the metabolic syndrome: acetyl-CoA carboxylase inhibition. Expert Opin Ther Targets. 2005; 9:267–81.
91. Loomba R, Kayali Z, Noureddin M, Ruane P, Lawitz EJ, Bennett M, Wang L, Harting E, Tarrant JM, McColgan BJ, Chung C, Ray AS, Subramanian GM, Myers RP, Middleton MS, Lai M, Charlton M, Harrison SA. GS-0976 reduces hepatic steatosis and fibrosis markers in patients with nonalcoholic fatty liver disease. Gastroenterology. 2018; 155:1463–73.
92. Barondes SH, Castronovo V, Cooper DN, Cummings RD, Drickamer K, Feizi T, Gitt MA, Hirabayashi J, Hughes C, Kasai K, Leffler H, Liu FT, Lotan R, Mercurio AM, Monsigny M, Pillai S, Poirer F, Raz A, Rigby PWJ, Rini JM, Wang JL. Galectins: a family of animal beta-galactoside-binding lectins. Cell. 1994; 76:597–8.
93. Ochieng J, Fridman R, Nangia-Makker P, Kleiner DE, Liotta LA, Stetler-Stevenson WG, Raz A. Galectin-3 is a novel substrate for human matrix metalloproteinases-2 and -9. Biochemistry. 1994; 33:14109–14.
94. Yang RY, Hsu DK, Liu FT. Expression of galectin-3 modulates T-cell growth and apoptosis. Proc Natl Acad Sci U S A. 1996; 93:6737–42.
95. Jeng KC, Frigeri LG, Liu FT. An endogenous lectin, galectin-3 (epsilon BP/Mac-2), potentiates IL-1 production by human monocytes. Immunol Lett. 1994; 42:113–6.
96. Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, Hirashima M, Liu FT. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000; 165:2156–64.
97. Tellez-Sanz R, Garcia-Fuentes L, Vargas-Berenguel A. Human galectin-3 selective and high affinity inhibitors. Present state and future perspectives. Curr Med Chem. 2013; 20:2979–90.
98. Harrison SA, Marri SR, Chalasani N, Kohli R, Aronstein W, Thompson GA, Irish W, Miles MV, Xanthakos SA, Lawitz E, Noureddin M, Schiano TD, Siddiqui M, Sanyal A, Neus-chwander-Tetri BA, Traber PG. Randomised clinical study: GR-MD-02, a galectin-3 inhibitor, vs. placebo in patients having non-alcoholic steatohepatitis with advanced fibrosis. Aliment Pharmacol Ther. 2016; 44:1183–98.
99. Chalasani N, Abdelmalek MF, Garcia-Tsao G, Vuppalanchi R, Alkhouri N, Rinella M, Noureddin M, Pyko M, Shiffman M, Sanyal A, Allgood A, Shlevin H, Horton R, Zomer E, Irish W, Goodman Z, Harrison SA, Traber PG; Belapectin (GRMD-02) Study Investigators. Effects of belapectin, an inhibitor of galectin-3, in patients with nonalcoholic steatohepatitis with cirrhosis and portal hypertension. Gastroenterology. 2020; 158:1334–45.
100. Pedrosa M, Seyedkazemi S, Francque S, Sanyal A, Rinella M, Charlton M, Loomba R, Ratziu V, Kochuparampil J, Fischer L, Vaidyanathan S, Anstee QM. A randomized, double-blind, multicenter, phase 2b study to evaluate the safety and efficacy of a combination of tropifexor and cenicriviroc in patients with nonalcoholic steatohepatitis and liver fibrosis: study design of the TANDEM trial. Contemp Clin Trials. 2020; 88:105889.


 PDF
PDF Citation
Citation Print
Print



 XML Download
XML Download