

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Propofol (2,6-diisopropylphenol) is a short-acting drug used in anesthesia induction and maintenance in short procedures and outpatient procedures [1]. When administered intravenously, it induces anesthesia at a rate similar to barbiturates, but post-operative recovery is much faster [2]. Propofol is one of the most preferred drugs in intensive care units because it is a strong anesthetic and effective sedative [3,4]. However, multiple organs and system damage known as propofol infusion syndrome (PIS) have been reported in those using propofol for a long time for sedation [5]. Retrospective studies have shown that the risk of PIS is high in all patients receiving short-term infusions at high doses or long-term infusions at different doses [5]. The literature has revealed that PIS progresses with severe metabolic acidosis, rhabdomyolysis, kidney, and heart failure, and can even lead to death [6]. One of the causes of mortality, rhabdomyolysis is a serious muscle injury characterized by the breakdown of skeletal muscle cells. It has been reported that skeletal muscle and myocardial cell damage markers such as creatine kinase (CK), creatine kinase MB (CK-MB), and troponin-I (TP I) levels are increased in PIS [7,8]. It has also been reported that markers of liver damage such as lactate dehydrogenase (LDH), aspartate aminotransferase (AST), alanine aminotransferase (ALT) are also increased in PIS [8]. It has been explained that one of the important clinical signs of PIS in literature is renal failure [9]. In histopathological examination, necrosis was observed in the renal tubules [10]. This event is followed by progressive increases in serum creatinine and blood urea nitrogen (BUN) concentrations [11]. Serum creatinine and BUN are used to evaluate oxidative kidney damage. The increase in creatinine and BUN has been associated with an increase in malondialdehyde (MDA), an oxidant parameter, and a decrease in endogenous antioxidant levels such as glutathione (GSH) [12]. A decrease in intracellular adenosine triphosphate (ATP) levels has been noted as the underlying cause of all forms of rhabdomyolysis [13]. Cray et al. [14] have shown that propofol has a destructive effect on the respiratory chain and leads to a decrease in ATP production [5]. An increase in myoplasmic Ca2+ concentration was detected in parallel with the decrease in intracellular ATP levels in rhabdomyolysis [13]. The increase of intracellular Ca2+ is caused by the decrease in ATP level due to ATPase inhibition [15]. It is known that increased intracellular Ca2+ ion concentration causes oxidative stress and leads to the initiation of pathological events in cells [16].
No data was found in the literature indicating oxidative stress in the pathogenesis of propofol-induced rhabdomyolysis. However, the pathophysiological process has been reported to include oxidative stress and inflammation in the glycerol-induced rhabdomyolysis (animal) model [17]. It has been noted that the damage caused by reactive oxygen species (ROS) such as hydrogen peroxide is associated with the loss of mitochondrial membrane potential and a decrease in ATP levels [18]. It has been reported that drugs increasing ATP levels restore mitochondrial damage and protect cells from ROS damage [19]. ATP to be examined in the present study for its possible protective effect against propofol-induced muscle damage is an organic compound involved in energy transformations in all living cells and contains carbon (C), hydrogen (H), oxygen (O), nitrogen (N) and phosphate (P). More than 95% of the molecular oxygen is used by mitochondria for energy generation in the form of ATP [20]. The energy released from ATP degradation is the primary energy source for muscle contraction [21]. Kumbasar et al. [22] have highlighted that exogenous ATP protects ischemic tissue from oxidative stress by inhibiting the excessive production of oxidant markers such as xanthine oxidase and malondialdehyde and the consumption of antioxidant total glutathione. It is understood from the literature that rhabdomyolysis is a clinical condition of skeletal muscle destruction. Also, it shows that using the therapeutic dose for a long time or in high doses for a short time may lead to PIS. The aim of our study is to biochemical and histopathologically investigate the protective effect of ATP against possible skeletal muscle damage in rats treated with propofol at a therapeutic dose for a long time and at high doses for a short time.
Go to : 
METHODS
Animals
A total of 30 male albino Wistar rats weighing 245–257 grams were used in the experiment. The animals were obtained from Ataturk University Medical Experimental Application and Research Center. The animals were housed and fed in groups in normal laboratory conditions (22˚C) before the experiment. Animal experiments were performed by the National Guidelines for the Use and Care of Laboratory Animals and were approved by the local animal ethics committee of Ataturk University, Erzurum, Turkey (Ethics Committee [Dated: 28/10/2019, Meeting no:88012460-840.01-E50907]).
Chemicals
Propofol (1%-20 ml ampoule) was obtained from Fresenius Kabi Pharmaceutical San-Turkey, thiopental sodium was provided by IE Ulagay-Turkey, and ATP was supplied by Zdorove Narodu Ukraine.
Experimental groups
Experimental animals were divided into five groups as healthy controls (HG), 50 mg/kg propofol (P-50), 50 mg/kg propofol + 4 mg/kg ATP (PA-50), 100 mg/kg propofol (P-100), and 100 mg/kg propofol + 4 mg/kg ATP (PA-100).
Experimental procedure
ATP was injected into the PA-50 (n = 6) group at a dose of 4 mg/kg intraperitoneally (IP) to experiment. An equal volume (0.5 ml) of distilled water was administered to the P-50 (n = 6) and HG (n = 6) groups simultaneously. One hour after the administration of ATP and distilled water, 50 mg/kg propofol was injected IP to P-50 and PA-50 groups. This procedure was performed once a day for 30 days.
Furthermore, 4 mg/kg ATP was IP injected to the PA-100 (n = 6) group and an equal volume of distilled water was administered to the P-100 (n = 6) group. After one hour, 100 mg/kg propofol was intraperitoneally injected into both groups. This procedure was performed three times with an interval of 1 days. At the end of this period, CK, CK-MB, creatinine, BUN, TP I, AST, ALT, and LDH were measured in blood samples taken from all animal groups. Also, MDA and total glutathione (tGSH) levels were measured in skeletal muscle tissue samples of animals killed by decapitation. Some of the skeletal muscle tissues were examined histopathologically. Biochemical and histopathological results obtained from all experimental groups were compared and evaluated.
Biochemical analyses
CK and CK-MB measurement: Analysis of plasma CK and CK-MB enzymes were measured using the commercial kit (11447378 122, Roche) in Cobas c701 system (Roche Molecular Diagnostics, Pleasanton, CA, USA).
BUN measurement: Quantitative determination of serum urea level was done by using the commercial kit (Cat. No. 10171778 122, Roche) in Cobas 8000 autoanalyzer by spectrophotometric method.
TP I assay: Troponin I (Troponin I kit - DRG Diagnostic) levels were measured in the ELISA device (ELISA reader® -DAS) using a commercial kit (EIA-2952).
ALT and AST analysis: AST and ALT values were determined in cobas integra kit (ALT, 20764957322), (AST, 20764949322) as enzymatic.
LDH analysis: Serum LDH enzyme activity measurement was determined using the Cobas Integra kit (LDH, 03004732122).
Biochemical analysis of the muscle tissue: Homogenates were prepared from tissues for biochemical analysis on liver tissues. tGSH and MDA levels in the supernatants obtained from these homogenates were determined using appropriate methods in the literature.
Preparation of samples: At this stage of the study was weighed 0.2 g of each tissue sample removed. For MDA measurement, 1.15% potassium chloride solution was completed to 2 ml in phosphate buffer pH = 7.5 for tGSH measurement and homogenized in ice medium. The tubes were then centrifuged at +4°C at 10,000 rpm for 15 min. Supernatants were used as the analysis sample.
MDA measurement: MDA measurement is based on the spectrophotometric measurement (at 532 nm) of absorbance of a pink colored complex formed by thiobarbituric acid and MDA at high temperature (95°C) [23].
tGSH measurement: DTNB (5,5’-Dithiobis [2-nitrobenzoic acid]) in the measurement medium is a disulfide chromogen and is readily reduced by compounds with sulfhydryl groups. The resulting yellow color was measured spectrophotometrically at 412 nm [24].
Histopathological examination: All of the tissue samples were first identified in a 10% formaldehyde solution for light microscope assessment. Following the identification process, tissue samples were washed under tap water in cassettes for 24 h. Samples were then treated with a conventional grade of alcohol (70%, 80%, 90%, and 100%) to remove the water within tissues. Tissues were then passed through xylol and embedded in paraffin. Four-to-five micron sections were cut from the paraffin blocks and hematoxylin-eosin staining was administered. Their photos were taken following the Olympus DP2-SAL firmware program (Olympus Inc., Tokyo, Japan) assessment. Histopathological assessment was carried out by the pathologist blind for the study groups. The severity of histopathological findings in each section was scored between grade 0–3 (0-normal, 1-mild damage, 2-moderate damage, and 3-severe damage).
Statistical analysis
For statistical analysis IBM SPSS 22 was used (released 2013. IBM SPSS Statistics for Windows, Version 22.0; IBM Corp., Armonk, NY, USA). The results were presented as mean ± standard deviation (SD). The distribution of variables was confirmed with the Shapiro–Wilk test. For biochemical measurements except CK-MB and Creatinine were compared with ANOVA. According to the homogeneity of variances, Tukey's HSD or Games-Howell tests were used as post-hoc test. For the comparisons of histopathological examinations, CK-MB and creatinine between groups Kruskal Wallis test was used with Dunn's test as a post-hoc test and adjusted p values were reported. A value of p less than 0.05 was considered significant for all tests.
Go to :
RESULTS
Biochemical findings
CK, CK-MB, BUN, and TP I analysis results: As can be seen from Fig. 1, CK activity significantly increased in the blood serum of animals treated with 50 mg/kg propofol for one month compared to healthy and ATP-treated groups (p < 0.001). Although the difference in CK activity between the 50 mg/kg propofol + ATP group and the healthy group was numerically close to each other, this difference was statistically significant (p < 0.01). Besides, propofol administered at a dose of 100 mg/kg increased CK activity further compared to the 50 mg/kg propofol group and this difference was significant compared to the healthy and ATP group (p < 0.0001). There was a significant difference in terms of CK activity between the propofol + ATP group and the healthy group (p < 0.05).

Similarly, 50 mg/kg propofol increased CK-MB level compared to the healthy group (p = 0.005) and the ATP group (p = 0.02), while at 100 mg/kg the difference was even more compared to the healthy group (p < 0.001) and the ATP group (p = 0.014). No difference was found in CK-MB activity between the 50 mg/kg propofol + ATP group and the healthy group (p = 0.25). Furthermore, there was no difference in CK-MB activity between the 100 mg/kg propofol + ATP group and the healthy group (p = 0.28).
Compared to the healthy group (p < 0.001) and the ATP group (p < 0.001), 50 mg/kg propofol administration increased BUN level significantly. BUN level increased even more significantly by 100 mg/kg propofol administration compared to the healthy group (p < 0.0001) and the ATP group (p < 0.0001). The difference in the BUN level between the 50 mg/kg propofol + ATP group and the healthy group was statistically insignificant (p = 0.45). However, the difference in the BUN level between the 100 mg/kg propofol + ATP group and the healthy group was statistically significant (p < 0.001).
Also, 100 mg/kg propofol administration increased serum TPI levels of animals more significantly compared to the dose of 50 mg/kg (p < 0.001). The difference in TPI levels between the healthy group and 50 mg/kg propofol + ATP group was statistically insignificant (p = 0.12). The difference in the TPI level between the 100 mg/kg propofol + ATP group and the healthy group was not found statistically significant (p = 0.07).
ALT, AST, and LDH analysis results: As can be seen from Fig. 2, ALT activity significantly increased in the blood serum of animals treated with 50 mg/kg propofol for one month compared to healthy and ATP-treated groups (p < 0.001). Besides, propofol administered at a dose of 100 mg/kg increased ALT activity further compared to the 50 mg/kg propofol group and this difference was significant compared to the healthy and ATP groups (p < 0.0001). The difference in ALT activity between the healthy group and the 50 mg/kg propofol + ATP group was statistically insignificant (p > 0.05). The difference in ALT activity between the 100 mg/kg propofol + ATP group and the healthy group was statistically significant (p < 0.05).

Similarly, 100 mg/kg propofol administration increased AST activity further (p < 0.001) compared to 50 mg/kg propofol administration. ATP significantly inhibited the propofol-related increase in AST activity at 50 mg/kg (p < 0.0001) and 100 mg/kg (p < 0.0001) doses. There was a significant difference in AST activity between the 50 mg/kg propofol + ATP group and the healthy group (p < 0.05). A higher difference was observed in AST activity between the 100 mg/kg propofol + ATP group and the healthy group (p < 0.01).
Propofol caused an increase in LDH activity in the blood serum of animals. 100 mg/kg propofol administration caused a higher increase in LDH activity compared to 50 mg/kg (p < 0.001). No difference was found between the 50 mg/kg propofol + ATP group and the healthy group (p = 0.26). The difference between the 100 mg/kg propofol + ATP group and the healthy group was statistically significant (p = 0.002).
MDA and tGSH analysis results: As shown in Fig. 3, the amount of MDA in the muscle tissue of the animals treated with 50 mg/kg propofol for one month significantly increased compared to healthy and ATP-treated groups (p < 0.001). MDA level was higher in the 100 mg/kg propofol group compared to the 50 mg/kg propofol group (p < 0.001). The difference in MDA amount in the muscle tissue of the healthy group and that of the 50 mg/kg propofol + ATP group was statistically insignificant (p = 0.69). The difference in the MDA amount between the 100 mg/kg propofol + ATP group and the healthy group was statistically significant (p < 0.05).

The 50 and 100 mg/kg doses of propofol increasing MDA caused a decrease in tGSH in muscle tissue. Propofol further reduced the amount of tGSH at a dose of 100 mg/kg compared to 50 mg/kg (p < 0.001). There was a significant difference between the amount of tGSH in the muscle tissue of the 50 mg/kg propofol + ATP group and the healthy group (p = 0.009). This difference was more pronounced between the 100 mg/kg propofol + ATP group and the healthy group (p < 0.001).
Histopathological findings
Normal muscle cell nuclei, muscle fibers, and blood vessels in the skeletal muscle tissue of the healthy animal group are shown in Fig. 4. In the 50 mg/kg propofol group, pale muscle fibers having a marked swelling and irregular appearance, conjunctive blood vessels, and polymorphonuclear cell infiltration were observed (Fig. 5A). Significant recovery and improvement were observed in the group treated with 50 mg/kg propofol + ATP. In this group, the muscle fibers were regularly aligned and their color was normal. The degenerative disruption in muscle fibers decreased and blood vessels were normal (Fig. 5B). Furthermore, irregularity in adjacent muscle fibers, degeneration coupled with disruptions in transverse lines of the muscle fibers, and some torn fibers were observed in the 100 mg/kg propofol group. When the overall tissue was evaluated, dilated and conjunctive blood vessels were observed and numerous polymorphonuclear cell infiltrations were seen around the blood vessels (Fig. 5C). In the 100 mg/kg propofol + ATP group, muscle fiber nuclei and the alignment of muscle fibers showed normal appearance, mild confirmation in the blood vessels, and very few polymorphonuclear cells around the vessels were observed (Fig. 5D).
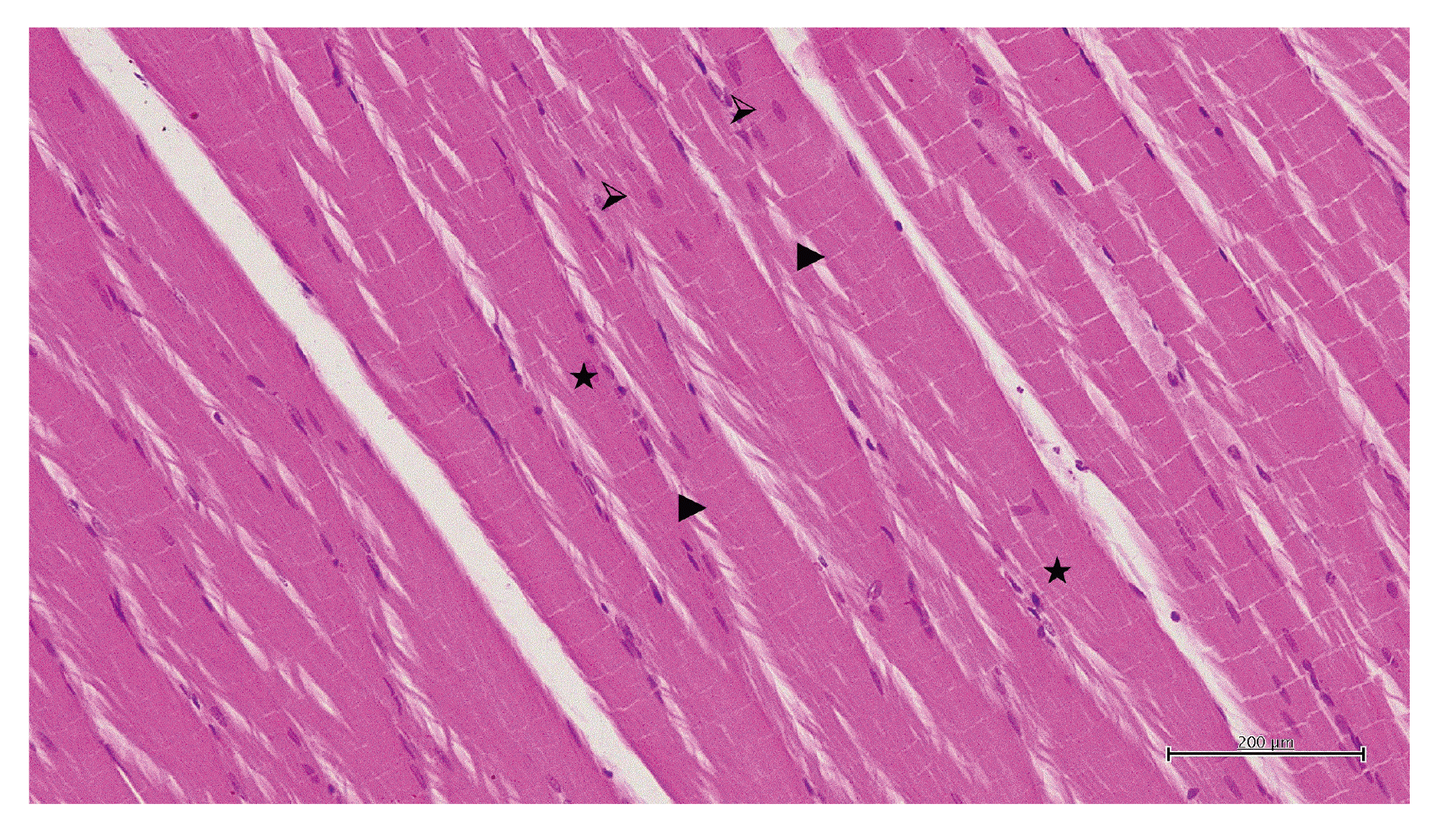 | Fig. 4Hematoxylin-eosin (H&E) staining in skeletal muscle tissue in the healthy group.
⮚, muscle cell nucleus; ►, muscle fibers; ★, blood vessel (H&E, ×200).
|
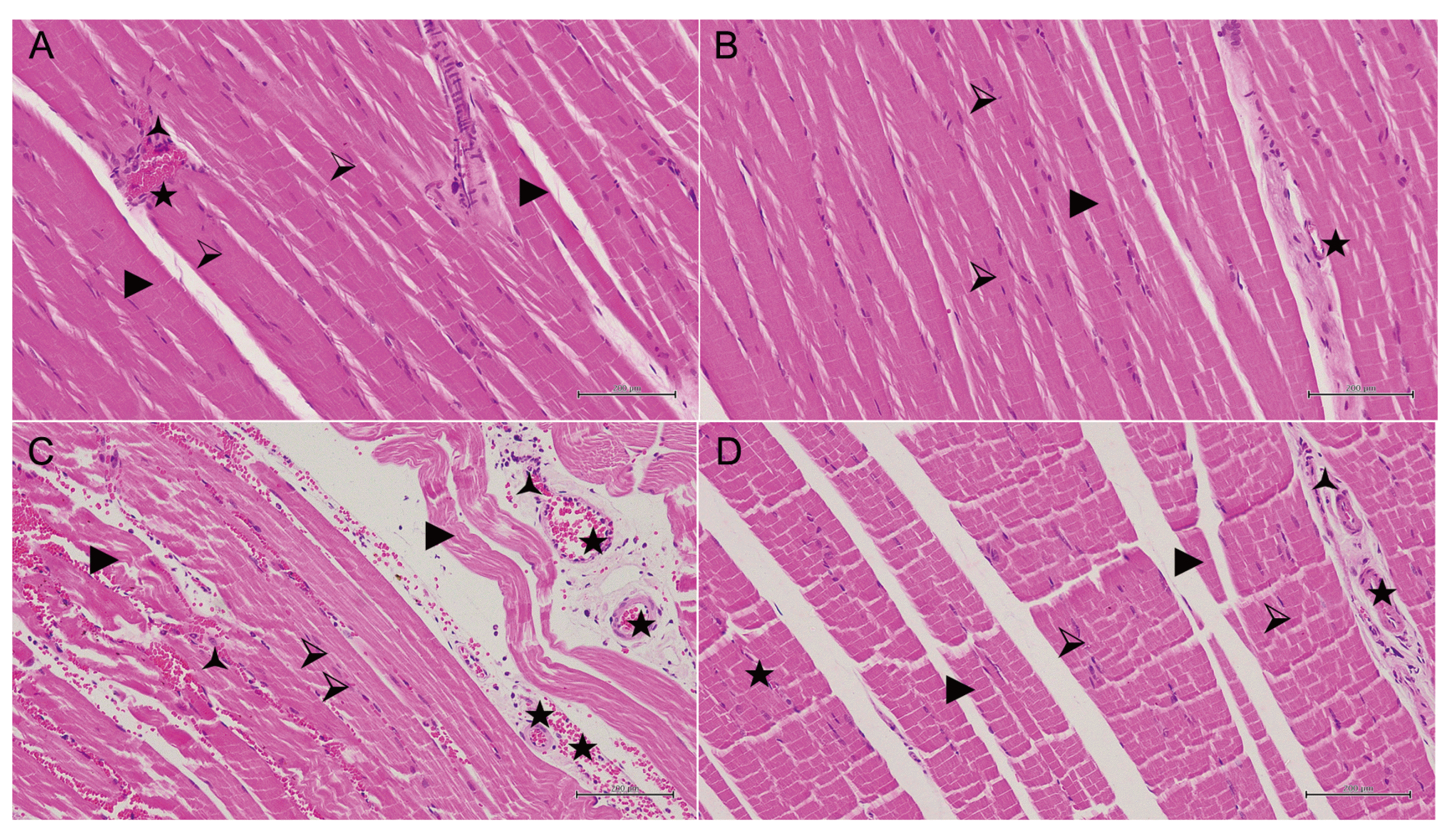 | Fig. 5Hematoxylin–eosin staining in skeletal muscle tissue in the low-dose (50 mg/kg) propofol group.(A) Hematoxylin-eosin (H&E) staining in skeletal muscle tissue in the low-dose (50 mg/kg) propofol group. ⮚, muscle cell nucleus; ►, degenerated pale muscle fibers with swollen, irregular appearance; ★, congested blood vessel; 🟀, polymorphonuclear cell infiltration (H&E, ×200). (B) H&E staining in skeletal muscle tissue in the low-dose (50 mg/kg) propofol + ATP group. ⮚, muscle cell nucleus; ►, decreased degeneration in muscle fibers with regular color and alignment; ★, normal blood vessel (H&E, ×200). (C) H&E staining in skeletal muscle tissue in the high-dose (100 mg/kg) propofol group. ⮚, disorganized and degenerated muscle cell nucleus; ►, irregular, ruptured and degenerated muscle fibers; ★, congested and dilated blood vessel; 🟀, numerously polymorphonuclear cell infiltration (H&E, ×200). (D) H&E staining in skeletal muscle tissue in the high-dose (100 mg/kg) propofol + ATP group. ⮚, normal muscle cell nucleus; ►, mostly normal organized and decreased degeneration in muscle fibers; 🟀, the spot of polymorphonuclear cell infiltration; ★, mildly congested blood vessel (H&E, ×200).
|
Go to :
DISCUSSION
In this study, the effect of ATP on the propofol administration at different doses, different intervals, and different durations was investigated biochemically and histopathologically. Our biochemical results showed that CK, CK-MB, TPI, ALT, AST, LDH, creatinine, and BUN levels were significantly higher in the blood serum of animals treated with 100 mg/kg propofol compared to 50 mg/kg. Besides, muscle MDA level was higher whereas tGSH was lower in the 100 mg/kg propofol group compared to 50 mg/kg. Recent studies have reported that high-dose short-term propofol infusion causes PIS in patients. Furthermore, it has been reported that PIS develops in all patients undergoing long-term infusion at a low dose range [5]. CK enzymes are used in the clinics to determine whether there is any damage to the skeletal and cardiac muscle tissue of individuals [25]. In the literature, it is stated that an increase in CK is associated with the extent of muscle damage and the severity of the disease. Furthermore, it is emphasized that muscle cell damage is caused by CK leak from the cells into the blood serum [26]. Therefore, measuring serum CK activity is an important indicator of muscle cell necrosis and all types of muscular dystrophy [27]. Another important parameter used in the diagnosis of myocardial infarction and inflammatory skeletal muscle diseases is the CK-MB isoenzyme [28]. Chronic necrotic muscle injury can lead to an increase in CK-MB activity [29]. CK-MB increases together with CK in patients with chronic kidney disease and destructive myopathies such as Duchenne muscular dystrophy, polymyositis, and dermatomyositis [30,31].
In our study, propofol increased the amount of serum TPI as well. In a previous study, it was reported that troponin increased in propofol-associated cardiac muscle damage [32]. TPI is known to be a highly sensitive parameter in myocardial damage [33]. Studies have suggested that the increase in TPI may be the result of a defect in the myocardia membrane caused by reactive oxygen products [34]. Propofol-associated ALT and AST elevations are multifactorial processes including skeletal muscle damage and hepatocellular damage [35]. Serum ALT and AST levels usually increase in rhabdomyolysis and these enzymes are produced by skeletal muscle. On the other hand, simultaneous liver disease has been shown to elevate the levels of these enzymes [36]. ALT is generally considered as a more specific marker for hepatocyte damage. However, there is also evidence for the production of ALT outside the liver [37]. AST is found in the liver, skeletal muscle, heart, kidneys, pancreas, and erythrocytes. Therefore, both hepatic inflammation and injury of another organ should be considered in the case of elevated serum AST levels [38]. Our experimental results are consistent with the results of the previous studies [36]. LHD, as well as ALT and AST levels, were elevated in animals treated with propofol. High LDH activity has been reported in skeletal muscle, heart, lung, liver, and kidney damage associated with propofol application [10]. In the study of El-Ganainy et al. [39], high levels of LDH were associated with cellular damage in muscle tissue. In another study, it has been emphasized that LDH increases in rhabdomyolysis and is associated with oxidative stress [40]. One of the important clinical symptoms of PIS is kidney failure [6]. The severe increase in BUN and creatinine levels shows that it is caused by permanent damage to kidney tubules [41]. It is stated that increases in serum BUN and creatinine concentrations are affected by nonrenal factors and are also indicators of excessive damage such as loss of functional nephrons [42]. Previous studies have also reported an excessive increase in BUN and creatinine levels in blood samples with high oxidant parameters [12]. Our experimental results show that animals treated with 100 mg/kg propofol have significantly higher MDA levels and lower tGSH levels in muscle tissue compared to 50 mg/kg propofol. This suggests that propofol shifts the oxidant-antioxidant balance in the muscle tissue in favor of oxidants. MDA is a cytotoxic product formed by peroxidation of cell membrane lipids with ROS. MDA leads to cellular damage by causing cross-linking and polymerization of membrane components [43]. However, they are neutralized by GSH and other enzymatic non-enzymatic antioxidant defense systems against overproduced ROS in healthy tissues [44]. To the best of our knowledge, no data is indicating oxidative stress in the pathogenesis of propofol-induced muscle damage in the literature. However, the pathophysiological process has been reported to include oxidative stress and inflammation in the glycerol-induced rhabdomyolysis animal model [17]. In the ATP group, CK, CK-MB, TP I, ALT, AST, LDH, BUN, creatinine, MDA, and tGSH levels were similar to the levels measured in the healthy group. In a previous study, intracellular ATP reduction was shown to be the underlying cause of rhabdomyolysis [13]. In rhabdomyolysis, an increase in myoplasmic Ca2+ concentration was found in parallel with the decrease in intracellular ATP levels [13]. An increase in intracellular Ca2+ level causes activation of intracellular proteolytic enzymes and leads to the disintegration of myocytes and the release of large amounts of potassium, aldolase, phosphate, myoglobin, CK, LDH, and AST from the cells [15]. Also, it is known that increased Ca2+ concentration leads to oxidative stress and the initiation of pathological events in the cells [16].
Kumbasar et al. [22] reported that externally provided ATP protected ischemic tissue from oxidative stress by inhibiting the overproduction of xanthine oxidase and MDA, and consumption of tGSH. However, the information on whether ATP could enter or exit the cell was not well known until a certain time. The extracellular and intracellular effects of ATP have been clarified by understanding the release of ATP as well as the uptake of cells [45]. This literature information supports our experimental results obtained by the intraperitoneal application of ATP. In another study, it was reported that injected ATP was converted to ADP, AMP, adenosine, and inorganic phosphate products, and these products entered cells and some of ADP and AMP were re-phosphorylated to ATP in cells [46]. In a recent study, it is understood that our idea about ATP is supported [47]. The fact that 100 mg/kg propofol administration caused more severe histopathological damage in skeletal muscle tissue compared to 50 mg/kg shows that our biochemical test results are consistent with our histopathological findings. While degeneration, muscle fiber irregularity, vascular congestion, and percutaneous nephrolithotripsy (PNL) infiltration were moderate in the animals treated with 50 mg/kg propofol, these histopathological findings were severe at a dose of 100 mg/kg. ATP significantly reduced the muscle damage induced by propofol administration at both doses. Literature data show that the spectrum and clinical significance of histopathological findings related to rhabdomyolysis remain uncertain [48]. Muscular dystrophies represent a clinically and genetically heterogeneous group of diseases characterized by progressive weakness and skeletal muscle degeneration [49]. ATP depletion, which appears to be the result of many causes of rhabdomyolysis, is associated with myocyte degeneration [9].
Prolonged use of propofol at low doses may cause muscle and tissue damage in other organs, while propofol use at higher doses may cause more severe organ and tissue damage in a shorter period. Based on the results of this study, it can be concluded that ATP better prevents muscle damage and organ dysfunction caused by 50 mg/kg propofol administration compared to the dose of 100 mg/kg. These experimental results suggest that ATP may be clinically useful in the treatment of propofol-induced rhabdomyolysis and multiple organ damage.
Go to :
 XML Download
XML Download