

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Arterial thrombosis (AT) is the development of a thrombus in an artery. It can affect the cardio-cerebrovascular system. AT is among the top major healthcare problems worldwide. It can be caused by trauma, erosion, or rupture of an atherosclerotic plaque, and by other events that damage the artery surface and lead to blood clotting. An insoluble blood clot becomes a thrombus and may lead to acute myocardial infarction, arterial embolism, ischemic stroke, and other cardiovascular diseases [1,2]. The incidence rates of these conditions are high, as are associated mortality and morbidity rates [3,4]. Drug therapy for the treatment and management of AT has limitations and complications; thus, more ideal treatments for AT are needed.
The coagulation cascade is initiated by tissue factor (TF) upon tissue damage in situations of hemostasis and pathological thrombosis [5-7]. TF, also known as coagulation factor III, is a 47-kD transmembrane glycoprotein. It is a key cofactor of activated coagulation factor VIIa, which forms a complex that activates the whole blood coagulation cascade [8]. In the extrinsic coagulation pathway initiated by TF, prothrombin is converted to thrombin, which causes cellular responses such as fibrin formation, platelet activation, and thrombus formation [9]. Under pathological conditions, TF is released mainly by monocytes, neutrophils, and vascular endothelial cells; this release initiates life-threatening thrombosis in the context of sepsis, tumors, and atherosclerosis [10,11]. TF expression is regulated primarily by the phosphoinositide 3-kinase (PI3K)/Akt, glycogen synthase kinase 3 beta (GSK3β), and nuclear factor (NF)-κB signaling pathways [5,12,13]. The PI3K/Akt pathway regulates TF expression negatively through site-specific phosphorylation of GSK3β, and TF is regulated positively via activation of the NF-κB pathway. Thus, inhibition of the TF pathway via modulation of these interrelated signaling pathways is an effective strategy for the prevention of AT [14].
Endothelial (vascular) dysfunction is an early correlate of coronary artery disease in humans. It is also found in the context of severe inflammatory conditions [15]. Numerous studies have shown that inflammation is associated with an increased risk of cardiovascular disease [16-18]. Furthermore, these studies have provided substantial evidence for close relationships among inflammation, reactive oxygen species (ROS), and redox signaling. Oxidative stress and inflammation are also implicated in platelet activation and thrombus development [19]. ROS have been reported to stimulate a procoagulant state via TF expression [20].
Apurinic/apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1, henceforth referred to as Ref-1) is a multifunctional protein that is secreted from stimulated cells and has multiple roles. In addition to its anti-inflammatory function, it functions as an apurinic/apyrimidinic endonuclease in the DNA base repair pathway [21]. It modulates redox status by controlling the cellular responses to oxidative stress via the reduction of ROS production [22]. Recombinant human Ref-1 protein has been shown to inhibit proinflammatory cytokines and inflammation induced by tumor necrosis factor alpha (TNF-β) or lipopolysaccharide in endothelial cells and mice [23-25]. As chronic vascular inflammation is a key factor in the pathogenesis of various vascular diseases and in inflammatory pathways that promote thrombosis [26], the regulation of inflammatory reactions in the vascular endothelium may be a target for the treatment of thrombosis. Ref-1 could be a target for the treatment of AT, given its anti-inflammatory and anti-oxidative properties. However, whether Ref-1 inhibits AT or regulates TF-related signaling pathways in arterial vascular tissues is not known. Thus, in this study, we investigated the effect of adenovirus-mediated Ref-1 overexpression on carotid artery thrombosis and TF expression in a mouse model of thrombosis induced by treatment with FeCl3 solution. The possible role of the PI3K/Akt-GSK3β–NF-κB signaling pathway in AT was explored.
Go to : 
METHODS
Animals
Male Sprague-Dawley rats (Samtako, Osan, Korea) aged 8–10 weeks with body weights of 250–280 g were used in this study. All experimental procedures adhered to the animal care and use policies of Chungnam National University (CNUH-017-P0069). The animals were housed in a standard environment with a 12/12-h light/dark cycle, a constant room temperature of 20°C–25°C, and 40%–60% humidity. Food and water were supplied ad libitum.
Chemicals
2,2,2-Tribromoethanol, urethane, 2-methyl-2-butanol, dihydroethidium (DHE), nitrosodisulfonate (NDS), diethylenetriaminepentaacetic acid, hydroethidine, heparin, and acetonitrile were purchased from Sigma-Aldrich (St. Louis, MO, USA). SurgLide transparent gel was purchased from Bio-Chem Laboratories Inc. (Los Angeles, CA, USA).
Adenoviral infection
Adenovirus encoding β-galactosidase (adβ-gal) and full-length APE1/Ref-1 (adRef-1) were prepared. The carotid artery was infected with adenovirus using a previously described method with little modification [27-29]. Briefly, the rats were anesthetized by intraperitoneal injection of 0.25 g/kg 2,2,2-tribromoethanol. Following exposure of the left carotid artery by incision, artery was temporarily ligated by double approximator microvascular clamp (S&T AG, Neuhausen, Switzerland). Isolated arterial segments were washed once with saline followed by infection with saline or 1 × 108 pfu of adβ-gal or adRef-1 adenovirus and were incubated for 20 min. After incubation, temporarily ligated segments were washed again with saline and blood flow was recovered by release of microvascular clamp. The midline incision was closed with sutures and subsequent experiments were performed 24 h later.
Carotid artery thrombosis and measurement of arterial blood flow
The FeCl3 thrombosis model was modified based on previous reports [30,31]. Briefly, rats were anesthetized by intraperitoneal injection of 1.2 g/kg urethane 24 h after adenoviral infection. The carotid arteries were isolated. A TS420 perivascular flow module and nanoprobes (Transonic Systems Inc., Ithaca, NY, USA) were used to measure arterial blood flow. The signals were recorded constantly by a PowerLab 8/35 device (AD Instruments Ltd., Dunedin, New Zealand). The cavity between the artery and probe was filled with transparent gel, and injury was induced to external surface of the artery by attaching 1 × 2 mm filter paper soaked with 60% FeCl3 solution for 5 min. The time to occlusion (TTO), defined as the time at which no blood flow had been detected for > 5 min, was determined. The recording time was limited to 60 min.
Antibodies and immunoblotting
The following antibodies were used: Anti–Ref-1, anti–intercellular adhesion molecule-1 (ICAM-1), anti–vascular cell adhesion molecule-1 (VCAM-1), anti–P-65, anti–T-65, and anti-TF antibodies from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti–total GSK3β antibody from Abcam (Cambridge, UK). Anti–β-actin, anti–P-GSK3β, anti–P-Akt and anti–T-Akt antibodies from Cell Signaling Technology (Danvers, MA, USA). Four hours after FeCl3 injury, the arterial segments were harvested for immunoblotting. The segments were homogenized with RIPA buffer and incubated at 4°C for 1 h. Then, the homogenates were centrifuged at 12,000 g, 4°C for 30 min. The supernatant was collected and 30 μg lysate was used for immunoblotting with the appropriate primary and secondary antibodies. The chemiluminescent signal was developed using SuperSignal West Pico or Femto substrate (Pierce Biotechnology, Rockford, IL, USA). Values were normalized to a β-actin loading control.
Hematoxylin and eosin staining
Hematoxylin and eosin (H&E) staining was performed following a standard procedure. Eight minutes after FeCl3 treatment, the arteries were fixed in 10% formalin for 24 h at 4°C, then washed and embedded in paraffin. The paraffinized segments were sectioned, deparaffinized, rehydrated, and stained with H&E. The sections were digitally imaged under a microscope (BA210 Series; Motic, Richmond, BC, Canada).
ROS measurement by high-performance liquid chromatography
2-hydroxyethidium (2-EOH) was used to measure arterial superoxide production. High-performance liquid chromatography (HPLC) was used to detect 2-EOH, which is the oxidative form of DHE. For the standard, 2-EOH was made from NDS using a previously described method [32]. The arterial segments were excised 5 min after FeCl3 injury and washed with Krebs-HEPES buffer. They were then incubated with 50 μM DHE at 37°C for 30 min, washed again with Krebs-HEPES buffer at 37°C for 1 h, and homogenized with acetonitrile. The samples were centrifuged at 12,000 g, 4°C for 30 min. The supernatant was used for the HPLC assay, and the precipitate was lysed with NaOH to measure the protein concentration. 2-EOH was measured using a 1290 series device (Agilent technologies, Santa Clara, CA, USA) with a 120 Sb-C18 Poroshell column, 580 nm emission, and 480 nm excitation. The composition of the mobile phase was 37% acetonitrile and 0.1% trifluoroacetic acid. The flow rate of the mobile phase was 0.5 ml/min.
Go to :
RESULTS
Ref-1 suppresses FeCl3-induced AT in rats
To induce Ref-1 overexpression in the carotid artery, adenovirus-mediated gene delivery was used. Left carotid artery was exposed and the adenovirus encoded with β-gal or Ref-1 was introduced into the artery. The artery was incubated with the adenovirus for 24 h, then denuded. The infection site was exposed to 60% FeCl3 solution for 5 min. The blood flow was measured for 1 h. Four hours after thrombus formation, the artery was harvested for further experiments (Fig. 1A). The expression of Ref-1 was significantly higher in the AdRef-1–treated group than in the Adβ-gal–treated group (Fig. 1B and C). This result confirms that Ref-1 adenovirus-mediated gene delivery resulted in overexpression of the Ref-1 protein in the carotid artery.
 | Fig. 1Redox factor-1 (Ref-1) suppresses FeCl3-induced arterial thrombosis formation.(A) Timeline of experimental thrombus induction. (B) The arteries were infected with adβ-gal or adRef-1 for 24 h. Thrombosis was induced for 5 min, followed by 4 h incubation. The arteries were then harvested and Western blotting was performed to confirm Ref-1 overexpression. β-actin was used as an internal control. (C) The expression level of Ref-1 was quantified by densitometric analysis using ImageJ software. (D) FeCl3-mediated injury was induced in the adenovirus-infected carotid arteries for 5 min, and arterial blood flow was monitored constantly using a flow module until it had ceased entirely for 5 min. (E) Statistical graph of the TTO data. (F) After adenovirus infection and FeCl3-mediated injury, the arterial segments were isolated and stained with H&E. Scale bar 200 μm. (G) Statistical data are presented as ratios of the thrombus area. All data are presented as means ± standard deviations of three independent experiments. *p < 0.05 vs. Adβ-gal + FeCl3 (n = 3/group).
|
Following Ref-1 overexpression, FeCl3-soaked filter paper was attached to the infection site to initiate AT. The blood flow was then measured until occlusion. The TTO was defined as the time required for blood flow to cease entirely for > 5 min. The average TTO after FeCl3 exposure was 20 min in the control (Adβ-gal–treated) group; occlusion was delayed significantly (TTO = ~80 min) or did not occur in the AdRef-1–treated group (Fig. 1D and E).
H&E staining showed that the endothelial surface of the carotid artery was arranged in an orderly manner, with no thrombus formation in the artery lumen or inflammatory cell infiltration in the sham group. Carotid artery thrombosis was observed as compact red and white thrombus formation with thrombosis and the vascular wall connected to form a visible platelet trabecular. Most vascular endothelial cells exhibited thrombosis with a vascular wall surrounded by inflammatory cell infiltration. Adenovirus treatment alone did not result in AT formation, with outcomes similar to those in the sham group (data not shown). However, Adβ-gal + FeCl3 treatment resulted in the formation of thrombi that occupied almost 80% of the artery lumen and interrupted blood flow. In contrast, lumen treated with AdRef-1 + FeCl3 showed 50% occlusion by thrombi with reduced number of vascular endothelial cells at the surface and white blood cell adhesion and infiltration (Fig. 1F and G).
Ref-1 modifies the Akt/GSK3β/NF-κB signaling pathway and TFPI, TF and thrombomodulin expression
The PI3K/Akt signaling pathway negatively regulates TF expression [33-35]. We sought to investigate whether Ref-1 could regulate TF via the PI3K/Akt pathway. Ref-1 overexpression significantly increased Akt phosphorylation and the phosphorylation of GSK3β, a major downstream regulator of Akt (Fig. 2A and B). These results indicate that Ref-1 inhibited GSK3β activation by triggering its phosphorylation at the serine 9 phosphorylation site, which is related to GSK3β inactivation. GSK3β activation is required for the regulation of NF-κB activation [5,36]. Thus, we next examined the phosphorylation of p65. Ref-1 overexpression inhibited p65 phosphorylation (Fig. 2C and D). It also reduced TF expression (Fig. 2C and D), confirming that Ref-1 inhibits TF expression through PI3K/Akt signaling via the suppression of GSK3β activity.
 | Fig. 2Effect of redox factor-1 (Ref-1) overexpression on the Akt/glycogen synthase kinase 3 beta (GSK3β)/nuclear factor (NF)-κB signaling pathway and the expression of TF pathway inhibitor (TFPI), tissue factor (TF) and thrombomodulin. The arteries were infected with adβ-gal or adRef-1 for 24 h. Thrombosis was induced for 5 min, followed by 4 h incubation.The arteries were harvested and Western blotting was performed to measure the phosphorylation of (A) Akt and GSK3β, (C) p65, and the protein expressions of (C) TF, (E) TFPI and thrombomodulin. The total forms of the proteins and GAPDH were used as internal controls. The expression levels of the proteins were quantified by densitometric analysis using ImageJ software (B, D), and (F). All data are presented as means ± standard deviations of three independent experiments. *p < 0.05 vs. Adβ-gal + FeCl3 (n = 3/group).
|
TF expression is mainly regulated by TF pathway inhibitor (TFPI). TFPI is a single-chain polypeptide, which can reversibly inhibit Factor Xa. While Xa is inhibited, the Xa-TFPI complex can subsequently also inhibit the Factor VIIa-TF complex. In addition, thrombomodulin is an integral membrane protein expressed on the surface of endothelial cells, which serves as a cofactor for thrombin. It reduces blood coagulation by converting thrombin to an anticoagulant enzyme from a procoagulant enzyme. It also protects endothelial cells and vasculature by depressing inflammatory injuries. Therefore, we measured the protein expressions of both TFPI and thrombomodulin in the thrombotic carotid arteries as well as examined the effect of Ref-1 overexpression. As shown in Fig. 2E and F, Ref-1 overexpression increases the protein expressions of TFPI and thrombomodulin that were reduced by FeCl3 treatment.
Ref-1 inhibits inflammatory molecule expression in the carotid artery
Endothelial cell activation has been associated with the initiation of inflammation. It is identified via the expression of adhesion/inflammatory molecules, such as VCAM-1 and ICAM-1. An increase in VCAM-1 and ICAM-1 surface protein expression leads to an increase in vascular inflammation and the initiation of atherosclerotic plaque formation [37,38]. Thus, we examined the effect of Ref-1 overexpression on the expression of VCAM-1 and ICAM-1 in thrombotic carotid arteries. FeCl3 treatment significantly elevated VCAM-1 and ICAM-1 protein expression in the Adβ-gal–treated group (Fig. 3A and B). As expected, the expression of VCAM-1 and ICAM-1 was lesser in the AdRef-1 + FeCl3–treated group. This result suggests that Ref-1 inhibits endothelial activation.
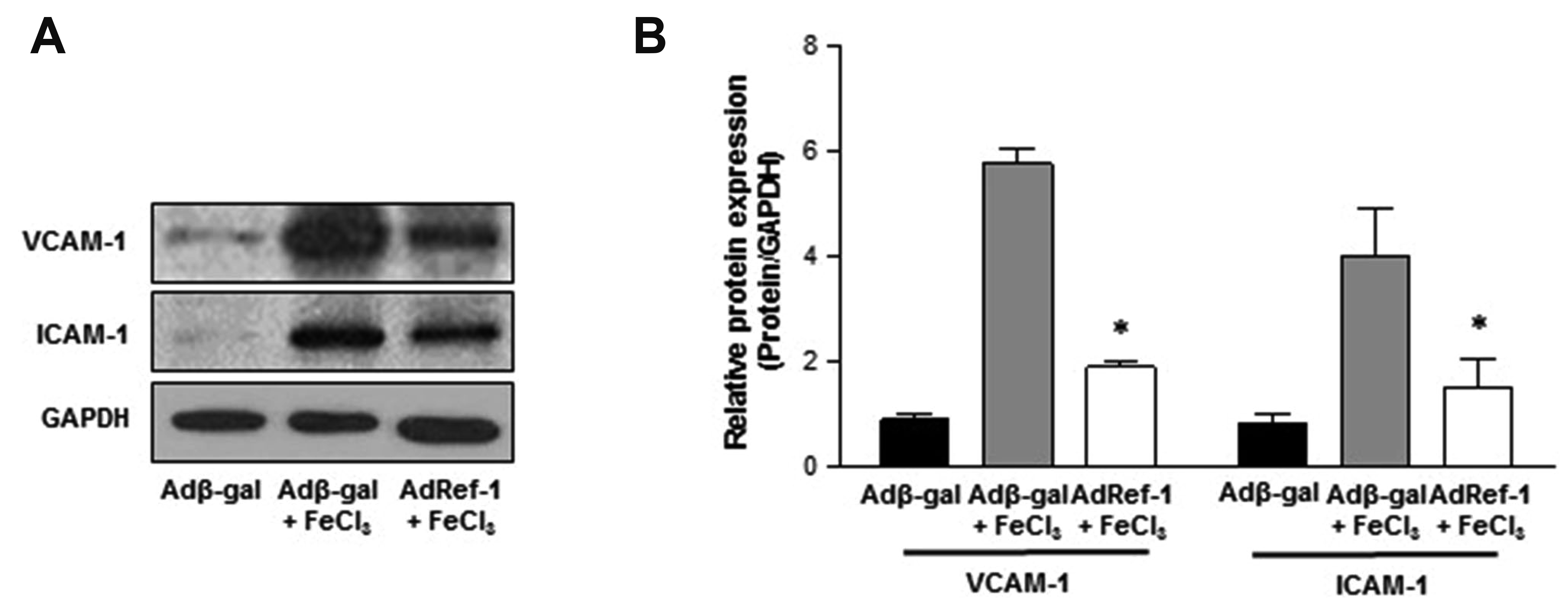 | Fig. 3Redox factor-1 (Ref-1) suppresses the expression of inflammatory molecules in the carotid artery.The arteries were infected with adβ-gal or adRef-1 for 24 h. Thrombosis was induced for 5 min, followed by 4 h incubation. The arteries were harvested and Western blotting was performed to measure the protein expressions of (A) vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1). GAPDH was used as an internal control. (B) The expression levels of VCAM-1 and ICAM-1 were quantified by densitometric analysis using ImageJ software. All data are presented as means ± standard deviations of three independent experiments. *p < 0.05 vs. Adβ-gal + FeCl3 (n = 3/group).
|
Ref-1 inhibits ROS generation in the carotid artery
Oxidative stress plays an important role in the development and progression of cardiovascular diseases. The majority of cardiovascular diseases are characterized by an imbalance between ROS formation and removal by cell antioxidant systems [39], which leads to the accumulation of superoxide, hydrogen peroxide, or peroxynitrite and results in loss of the steady state [40]. ROS participate directly in the regulation of platelet activation and thrombus formation [41]. Ref-1 is well known for its redox activity and antioxidant function. Thus, we measured ROS levels by quantifying the degree of DHE staining in arterial segments after FeCl3 injury with or without Ref-1 overexpression. DHE can freely permeate cell membrane and be oxidized by cellular O2•- to produce two red fluorescent products, namely ethidium, which is typically formed by a non-specific redox reaction, and 2-EOH, a specific adduct of cellular O2•-. 2-EOH can be readily separated from its parent DHE by performing HPLC and the 2-EOH peak can then be used to estimate the intercellular production of O2•-. As shown in Fig. 4A and B, the 2-EOH peak was considerably reduced by Ref-1 overexpression.
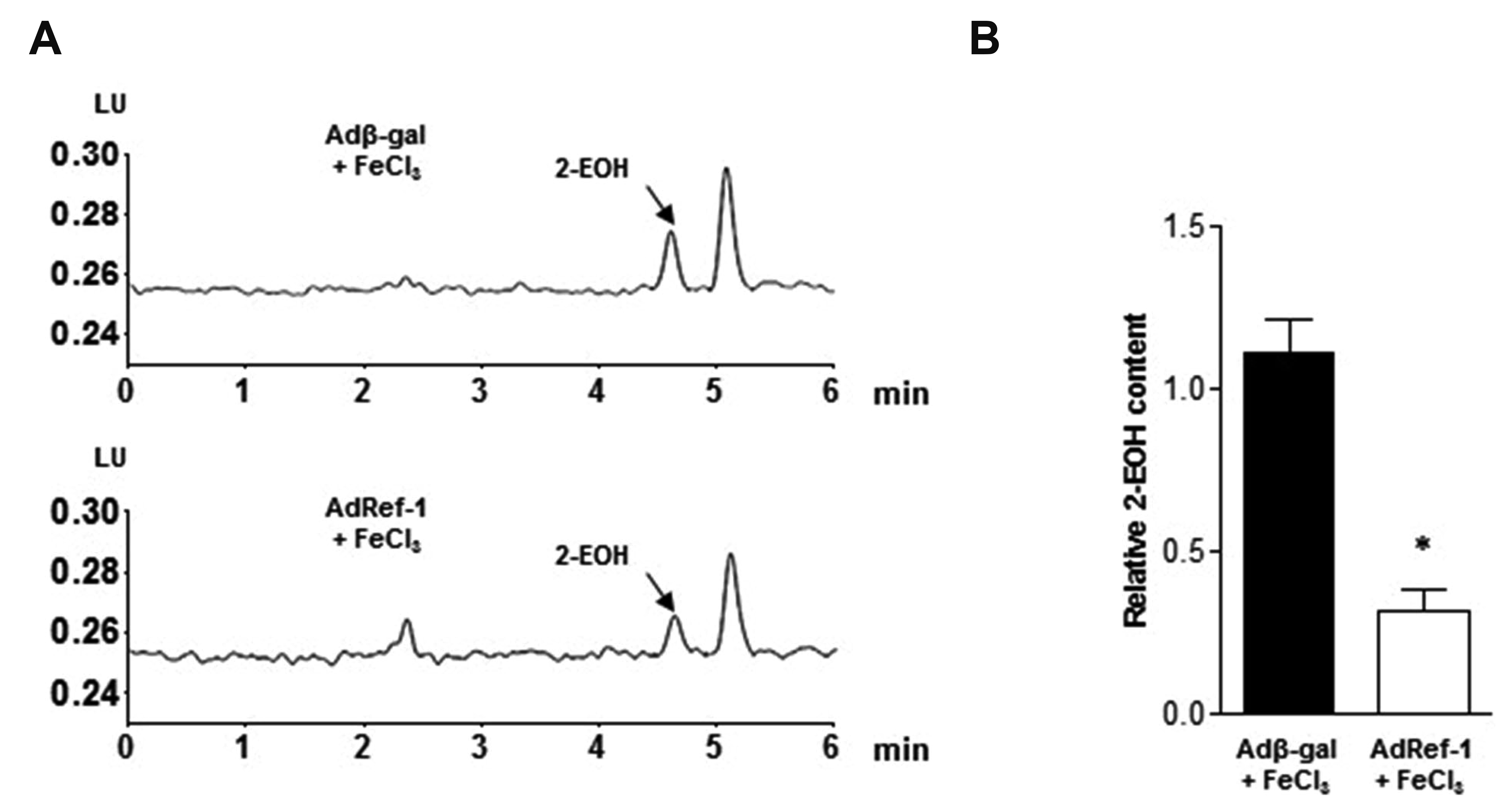 | Fig. 4Redox factor-1 (Ref-1) suppresses reactive oxygen species (ROS) production in the carotid artery.Arterial segments were isolated and stained with dihydroethidium (DHE) after adenovirus infection and FeCl3-mediated injury. (A) The fluorescent signal of 2-hydroxyethidium (2-EOH, the oxidative form of DHE) was measured by high-performance liquid chromatography to detect ROS levels. Arrows indicate the 2-EOH signals. (B) Statistical graph of the 2-EOH signal. All data are presented as means ± standard deviations of three independent experiments. *p < 0.05 vs. Adβ-gal + FeCl3 (n = 3/group).
|
Go to :
DISCUSSION
Vascular endothelial injury caused by inflammation is a key factor in the initiation of atherosclerosis and AT, as demonstrated in numerous studies [42-44]. Despite much research, however, the pathological mechanisms of AT are not completely understood, resulting in a lack of clinically safe and effective drug treatments. Under normal physiological conditions, vascular endothelial cells inhibit platelet aggregation, stimulate fibrinolysis, and inhibit abnormal coagulation. However, vascular endothelial cell activation stimulates platelet aggregation and inhibits the dissolution of fibrin, ultimately leading to abnormal luminal coagulation and thrombus formation [44]. To prevent and cure AT, the regulation of endothelial function, improvement of the coagulation balance, and alleviation of inflammation are thus essential. In this study, we used FeCl3 to induce carotid artery thrombosis. This model causes severe endothelial cell injury due to oxidative stress and increases TF expression in endothelial cells and leukocytes [45]. Thrombus formation achieved by this method is similar to clinical spontaneous thrombosis. Thus, this model has been used widely in the preclinical evaluation of thrombolytic drugs and related research [46].
Ref-1 is a protein with multiple functions. In addition to base-excision DNA repair and the transcriptional regulation of gene expression, it plays a significant role in the control of the cellular response to oxidative stress. In particular, Ref-1 reduces the intracellular production of ROS [47-49]. Ref-1 overexpression has been shown to suppress monocyte adhesion to endothelial cells induced by TNF-α [50] and to inhibit hypoxia-induced apoptosis of endothelial cells [51]. In addition, Ref-1 inhibited neointimal formation caused by balloon injury in rats [52]. These findings suggest that Ref-1 plays a protective role in the context of inflammatory vascular disease. However, no report has indicated that Ref-1 inhibits AT or modulates TF-related signaling pathways in arterial vascular tissues or endothelium. In this study, Ref-1 significantly improved blood flow in the carotid artery, delayed occlusion, and alleviated thrombosis, which is the first evidence that Ref-1 can inhibit AT (Fig. 1D–G).
Our findings demonstrate that the inhibition of TF expression in endothelial cells by Ref-1 occurs through the activation of TFPI, thrombomodulin, Akt/GSK3β signaling pathway and inhibition of the NF-κB signaling pathway (Fig. 2A–F). TF expression has been shown to be regulated primarily by the TFPI, NF-κB and PI3K/Akt signaling pathways [12,13,53,54]. NF-κB is a homo- or heterodimeric complex of proteins. The p65 heterodimer is involved primarily in the regulation of TF gene expression. In non-stimulated conditions, inactive NF-κB is localized in the cytoplasm, bound to IκB. Upon NF-κB activation, it dissociates from IκB, and the subsequent degradation of IκB proteins causes NF-κB dimers to enter the nucleus and induce TF gene expression [55]. The PI3K/Akt pathway is composed of a family of signal transduction enzymes that participate in the regulation of cellular functions such as cell proliferation and survival. PI3K/Akt is known to be a negative-feedback regulator of TF expression [34]. In addition, TFPI is a potent anticoagulant protein in the extrinsic coagulation pathway. It is a major physiological inhibitor of the coagulation initiation phase, acting as an inhibitor of TF-FVIIa complex [56]. Therefore, FeCl3 induced inhibition of TFPI in our study would allow coagulation process and enhancement of arterial thrombosis generation. Ref-1 overexpression increases TFPI protein levels that could in turn reduce TF-induced coagulation (Fig. 2E and F). Furthermore, thrombomodulin that is present on the endothelium throughout the vascular system is a vasculoprotective integral membrane glycoprotein, which is a crucial determinant of thromboresistance of ECs through anti-fibrinolytic, anti-coagulant, and anti-inflammatory effects [57]. It plays a central role in protection against excessive coagulation via activation of protein C and subsequent inactivation of coagulation factors V and VIII. FeCl3 injury reduces thrombomodulin expression. However, thrombomodulin increment following Ref-1 overexpression could be protective in terms of thrombotic events (Fig. 2E and F).
Previous studies have shown that the PI3K/Akt/NF-κB transduction pathways play a critical role in the induction of VCAM-1 and ICAM-1 expression [37]. Endothelial adhesion molecules such as VCAM-1 and ICAM-1, together with other important factors such as chemoattractants, play crucial roles in the homing of monocytes to vascular lesion sites. Significant modulation of ICAM-1 expression over time was observed in apoE-/- mice compared with that in wild-type mice [38]. Similarly, VCAM-1 expression has been identified as a very early event in the development of atherosclerotic lesions in experimental animal models [58]. In this study, Ref-1 abrogated the increased expression of VCAM-1 and ICAM-1 induced by FeCl3 injury (Fig. 3).
ROS are highly reactive metabolites that can damage various cellular constituents and are generated in vivo in several pathological conditions [59-61]. Many studies have demonstrated that ROS plays a pivotal role in the development of cardiovascular pathophysiology. Specifically, the restoration of blood flow after a period of ischemia was shown to generate an ROS surge [62]. Interestingly, ROS was also shown to promote thrombosis by inducing TF expression [60,63]. In this study, FeCl3 caused an increase in the ROS level, which was attenuated by Ref-1 (Fig. 4).
Taken together, the findings of this study reveal that Ref-1 plays a key role in the modulation of TF expression via the Akt/GSK3β/NF-κB signaling pathway, TFPI and thrombomodulin (Fig. 5). Ref-1 increases blood flow, reduces thrombus formation, and reduces oxidative stress in the carotid artery. These results provide evidence that Ref-1 plays an important role in AT. The present study might not be a confirmative but rather a preliminary study for the possibility that Ref-1 may be a new target for the treatment of AT and many other thrombotic diseases.
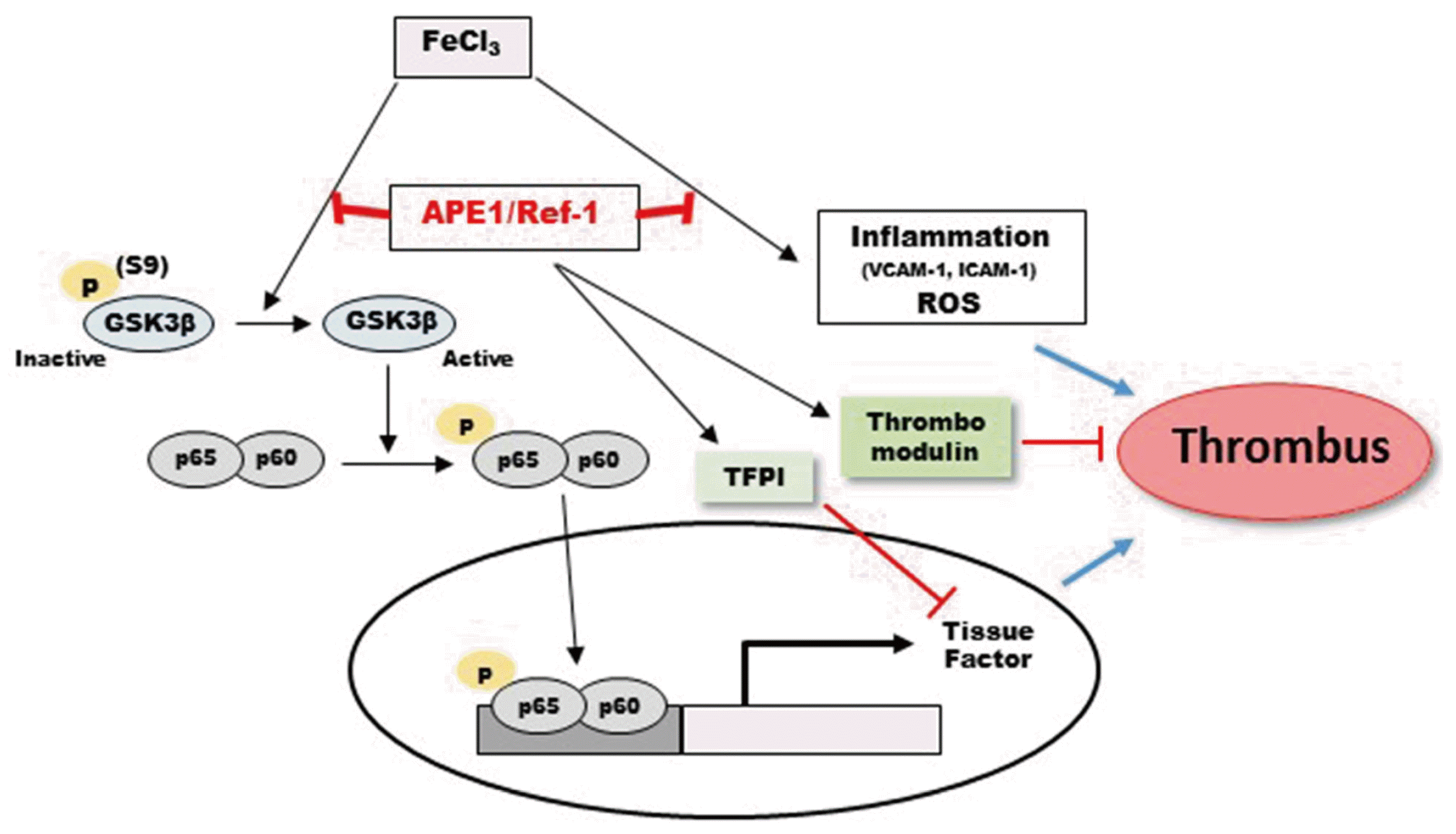 | Fig. 5Schematic model of the pathways involved in FeCl3-induced thrombus formation and the possible role of apyrimidinic endonuclease 1/redox factor-1 (APE1/Ref-1) in the inhibition of thrombus formation.GSK3β, glycogen synthase kinase 3 beta; TF, tissue factor; TFPI, TF pathway inhibitor; ROS, reactive oxygen species.
|
Go to :
 XML Download
XML Download