

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Neurodegenerative diseases are caused by neuronal cell death and rapid neuronal dedifferentiation processes, which result in severe nervous system dysfunction. For several decades, many research groups have investigated methods to prevent these diseases; during these investigations, the researchers found that neuronal cell death in these diseases is associated with diverse factors, including hypoxia, inflammation, protein misfolding, mitochondrial dysfunction, ethanol exposure, amyloid β, and both autophagy-mediated and proteasome-mediated protein degradation [1-6]. However, a common underlying factor in these diseases is the presence of aberrant levels of endogenous reactive oxygen species (ROS), which indiscriminately attack cellular components, leading to a variety of consequences. Thus, maintaining appropriate endogenous ROS levels is important for prevention of neurodegenerative diseases.
Changes in gene expression patterns due to epigenetic modifications play a crucial role in the progression of cellular differentiation, which may partly explain the differential gene expression of various types of cells [7,8]. Epigenetic changes are also associated with many types of diseases, including neurodegenerative diseases [9,10]. Recent studies have shown that aberrant repressor element-1 silencing transcription factor (REST)-dependent epigenetic changes are related to ischemia-induced neuronal cell death [11]. During neurodegeneration induced by chronic exposure to hypoxia or ethanol, neurons undergo epigenetic changes and eventually die [12,13]. Thus, researchers have focused on elucidating aberrant epigenetic changes that occur in a variety of diseases.
Histone methyltransferase G9a, a component of the REST complex, binds to a repressor element-1 (RE-1) consensus sequence [14,15]. This interaction of G9a with the RE-1 region leads to dimethylation of the lysine 9 residue of histone H3 (H3K9me2), which is associated with repression of RE-1-containing neuron-specific genes [16,17]. Aberrant G9a activation due to hypoxia or ethanol exposure increases the risk of neurodegeneration [18,19]. Targeted inhibition of G9a by a specific inhibitor [20] (e.g., BIX01294) has beneficial effects on anti-senescence [21] and promotes neuronal differentiation of mesenchymal stem cells [22]; thus, G9a inhibition may positively influence neurodegenerative disorders.
In this study, we examined the functional role of G9a in oxidative stress-induced neurodegeneration. We found that aberrant activation of G9a causes epigenetic disruption of the expression of neuron-specific genes in human neuroblastoma cells exposed to H2O2 (12 h), thereby leading to neurodegeneration. Furthermore, the G9a inhibitor BIX01294 attenuates this neurodegeneration.
METHODS
Cell culture
The SH-SY5Y human neuroblastoma cell line was purchased from KCLB (Korean Cell Line Bank, Seoul, Korea). SH-SY5Y cells were cultured in a 100-mm dish (SPL, Pocheon, Korea) in accordance with the manufacturer’s recommendations.
3(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay
Cell viability of BIX01294-treated cells was evaluated using the MTT assay (Sigma-Aldrich, St. Louis, MO, USA), in accordance with the manufacturer’s instructions. Briefly, 2 × 103 SH-SY5Y neuroblastoma cells were seeded onto 96-well plates. The next day, cells were incubated with 0–2 μM of BIX01294 for 12 h or exposed to H2O2 and then subjected to the MTT assay.
Quantitative polymerase chain reaction (real-time PCR)
Total RNA was isolated from SH-SY5Y cells using RNAiso reagent (TAKARA, Shiga, Japan), in accordance with the manufacturer’s instructions. Three micrograms of total RNA were used for reverse transcription with the AMPIGENE cDNA Synthesis Kit (Enzo, Farmingdale, NY, USA). cDNA was amplified using the Power SYBR Green PCR Master Mix (Applied Biosystems Inc., Foster City, CA, USA) with gene-specific primers for human MAP-2, NF-M, NEFH, SYN1, SCN3A, SNAP25, CACNA1A, and β-actin. The real-time PCR cycling parameters were as follows: 95°C for 10 min, followed by 40 cycles of 15 sec at 95°C and 1 min at 60°C. The primers used in this study were synthesized by GenoTech (GenoTech Corp., Daejeon, Korea) and IDT (Integrated DNA Technologies Inc., Coralville, IA, USA). The primers are summarized in Supplementary Table 1.
Chromatin immunoprecipitation
SH-SY5Y cells were incubated with or without 1 μM BIX01294 for 12 h, then treated with 100 μM H2O2 for 12 h. H3K9me2-containing chromatin was precipitated by an anti-H3K9me2 antibody (Merck, Darmstadt, Germany) and subjected to chromatin immunoprecipitation using the ChIP Assay Kit (Merck), in accordance with the manufacturer’s instructions. The details of this method were described in our previous study [22]. For real-time PCR analysis, 3 μl DNA was used for each reaction with specific primers for the proximal region of genomic DNA in human NF-M, SYN1, SNAP-25, and MAP-2 genes. The primer sequences are summarized in Supplementary Table 2.
Immunoblotting
Cells were incubated with 100 μl of radioimmunoprecipitation assay (RIPA) buffer containing protease and dephosphatase inhibitors (Santa Cruz Biotechnology, Dallas, TX, USA) for 30 min at 4°C and then centrifuged at 16,000 × g for 20 min. Total protein was then subjected to immunoblotting using antibodies against Cleaved caspase 3 (1:500), H3K9me2 (1:200; Merk), Histone 3 (1:1,000; Merk), Nestin (1:500), NF-M (1:500), MAP-2 (1:500), GAPDH (1:1,000; Santa Cruz Biotechnology), or β-actin (1:5,000; Sigma-Aldrich), followed by the appropriate horseradish peroxidase-conjugated secondary antibodies (1:10,000; Jackson ImmunoResearch Laboratories, West Grove, PA, USA). Details of primary and secondary antibodies are summarized in Supplementary Tables 3 and 4.
Neuronal differentiation
SH-SY5Y cells were incubated with or without 1 μM BIX01294 for 12 h (or 7 nM UCN0321 for 12 h). Then, cells were washed with phosphate-buffered saline (PBS) and treated with 100 μM H2O2 for 12 h. The cells were washed twice with PBS and returned to neuronal induction medium (Dulbecco’s Modified Eagle Medium-F12, 0.1% fetal bovine serum [FBS], and 1 μM retinoic acid) for 3–7 days. Control SH-SY5Y cells were incubated in 10% FBS medium for 3 days. Images were captured with a digital camera (i-Solution IMTcam3; Canon, Tokyo, Japan) under a microscope (Nikon ECLIPSE TS100; Nikon, Tokyo, Japan).
RESULTS
BIX01294 is protective against oxidative stress-induced apoptosis in neuronal cells
To examine the toxicity of BIX01294, SH-SY5Y cells were incubated with 0–2 μM of BIX01294 for 12 h. No cytotoxicity was observed with ≤ 1.5 μM BIX01294, whereas viability decreased by approximately 10% on exposure to 2 μM BIX01294 (Fig. 1A). To estimate cell viability in H2O2-treated cells, SH-SY5Y cells were treated with 0–100 μM H2O2 for 12 h. Survival rates decreased in a dose-dependent manner (Fig. 1B) and reached 60% in 100 μM H2O2-treated cells.

Fig. 1
Protective effects of BIX01294 in oxidative stress-damaged SH-SY5Y cells.
(A) Toxicity was measured by the MTT assay in SH-SY5Y cells that were treated with 0–2 μM BIX01294 (t-test, *p < 0.05, mean ± SD, n = 3). (B) SH-SY5Y cells were treated with 0–100 μM H2O2 for 12 h (t-test, *p < 0.05, **p < 0.005, mean ± SD, n = 3). (C) SH-SY5Y cells were incubated with 0–1.5 μM BIX01294, then treated with or without 100 μM H2O2 (t-test, ##p, **p < 0.005, mean ± SD, n = 3). (D) SH-SY5Y cells were treated with 1 μM BIX01294 for 0–24 h, then treated with or without 100 μM H2O2 for 12 h (t-test, #p < 0.05, **p < 0.005, mean ± SD, n = 3). (E, F) Immunoblot analysis was performed with antibodies specific to cleaved caspase-3 in H2O2-treated SH-SY5Y cells, with or without BIX01294 treatment (t-test, *p < 0.05, mean ± SD, n = 3). Ctrl indicates control cells, which were not treated with BIX01294 or H2O2. BIX indicates BIX01294.
![]()
To examine the protective effect of BIX01294 in oxidative stress-induced neuronal cells, SH-SY5Y cells were incubated with 0–1.5 μM BIX01294 for 12 h (Fig. 1C) or 1 μM BIX01294 for 0–24 h (Fig. 1D). Cells were then exposed to 100 μM H2O2 for 12 h. Maximal protective effects were observed when cells were incubated with 1 μM BIX01294 for 12 h (Fig. 1C, D). These effects were confirmed by immunoblot analysis with apoptotic marker cleaved caspase-3. Cleaved caspase-3 levels increased when cells were exposed to H2O2, but not in cells that were pre-incubated with 1 μM BIX01294 (Fig. 1E). The protein expression levels were quantified (Fig. 1F). These results suggest that BIX01294 treatment prevents cell damage by oxidative stress.
Oxidative stress-induced reduction in the expression of neuron-specific genes was attenuated by G9a inhibition
To further investigate the influence of H2O2 on neuronal cells, SH-SY5Y cells were treated with or without 1 μM BIX01294 for 12 h, then treated with 100 μM H2O2 for 12 h. Gene expression was measured by real-time PCR with primers for neuron-specific genes: NF-M, NEFH, MAP-2, SCN3A, CACNA1A, Syn1, and SNAP25. As shown in Fig. 2A, expression of the tested genes decreased in H2O2-treated cells; however, expression levels of NF-M, NEFH, MAP-2, CACNA1A, and SYN1 showed significant recovery on treatment with BIX01294. Immunoblot analysis was performed with antibodies against neuron-specific proteins (Nestin, NF-M, and MAP-2); this analysis showed that protein levels recovered on treatment with BIX01294 (Fig. 2B). These findings indicate that neuron-specific markers decreased upon exposure to H2O2-induced oxidative stress, and that the expression levels of these markers were restored on treatment with BIX01294.

Fig. 2
Oxidative stress-induced reduction in the expression of neuron-specific genes was partially restored by G9a inhibition.
(A) The expression levels of neuron-specific genes in H2O2-treated SH-SY5Y cells (H2O2) and in SH-SY5Y cells treated with both H2O2 and BIX01294 (BIX-H2O2) were measured by real-time PCR; these expression levels were compared with those in SH-SY5Y cells in normal conditions (Ctrl; t-test, #p, *p < 0.05, ##p, **p < 0.005, mean ± SD, n = 4). *p, **p (vs. control), #p, ##p (vs. 100 μM H2O2-treated cells). (B) The neuron-specific proteins Nestin, NF-M, and MAP-2 were examined by immunoblot analysis in H2O2-treated SH-SY5Y cells with or without BIX01294 treatment. (C) Protein expression levels were quantified using ImageJ software. β-actin was used as the internal standard.
![]()
Oxidative stress-induced downregulation of RE-1-containing neuronal genes was mediated by aberrant activation of G9a
The G9a inhibitor BIX01294 can partially prevent cell damage induced by oxidative stress; thus, we next examined the relationship between oxidative stress and G9a. Activated G9a dimethylates the lysine 9 residue of histone H3 (H3K9), thereby producing dimethyl-H3K9 (H3K9me2) [23]. We performed immunoblot analysis using the H3K9me2 antibody to assess G9a activity. H3K9me2 was increased 55.6% (1.8-fold) in H2O2-treated cells (vs. control cells) and decreased by as much as 17.7% with BIX01294 treatment (vs. H2O2-treated cells) (Fig. 3A). To further examine these effects in H2O2- and BIX01294-treated cells, chromatin immunoprecipitation (ChIP) assays for H3K9me2 were performed with RE-1 site-containing neuron-specific genes. H3K9me2 occupancy of the RE-1 site of the neuron-specific genes NF-M, Syn1, SNAP-25, and Map-2 was significantly increased on exposure of cells to H2O2, whereas it was significantly decreased on treatment with BIX01294 (significant decrease for NF-M, SNAP-25, and Map-2; no significant difference for Syn1; Fig. 3B). These results indicate that oxidative stress induces aberrant activation of G9a, thereby disrupting neuron-specific gene expression.
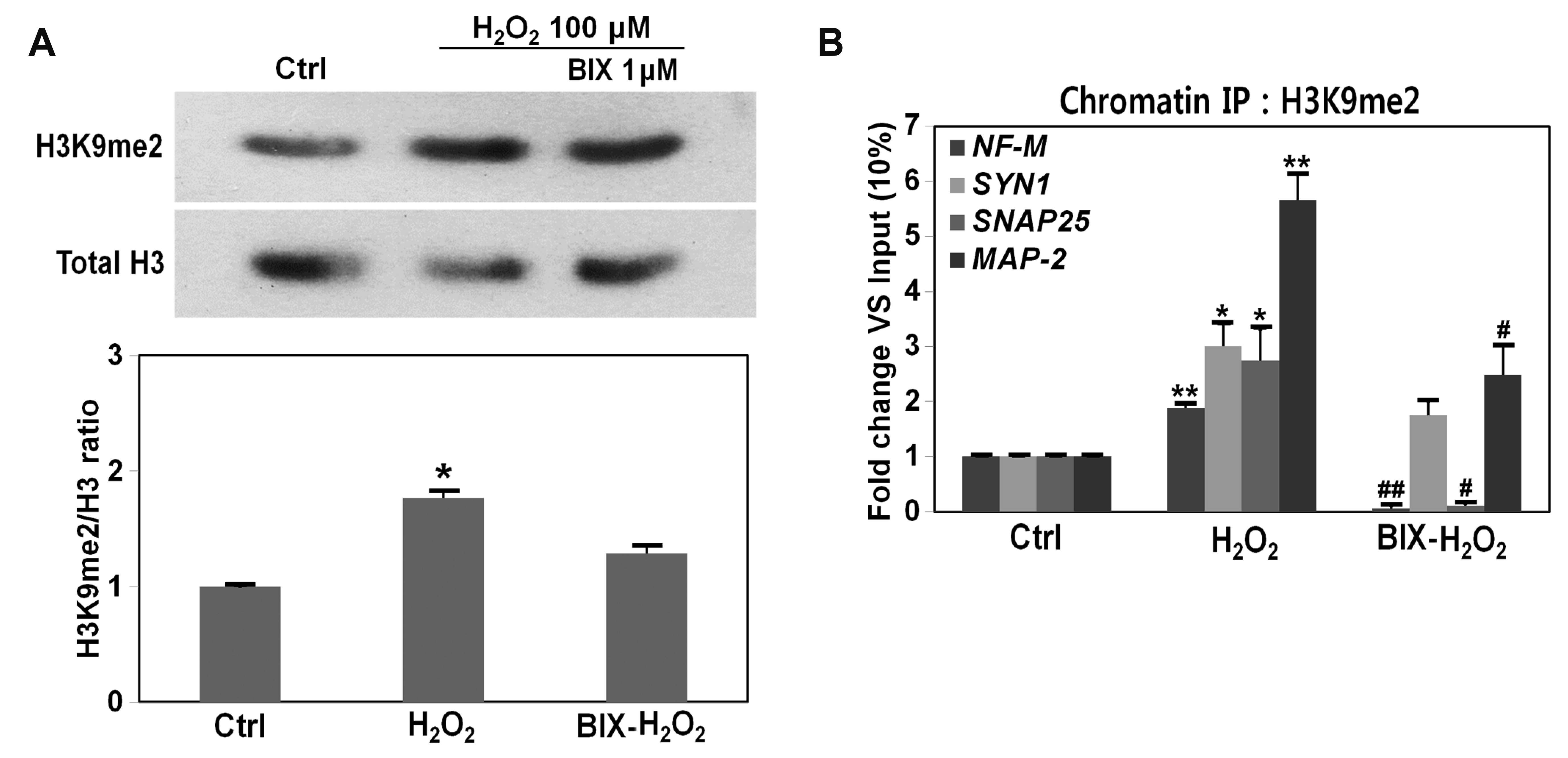
Fig. 3
G9a is aberrantly activated by oxidative stress and disrupts the expression of RE-1-containing neuron-specific genes.
(A) G9a activity was measured by immunoblot analysis with an antibody specific for H3K9me2 (top panel). The ratio of H3K9me2:H3 expression was measured using ImageJ software (bottom panel) (t-test, *p < 0.05, mean ± SD, n = 4; vs. control). (B) For the ChIP assay, SH-SY5Y cells treated with H2O2, with or without BIX01294 treatment (indicated by H2O2 or BIX-H2O2), were immunoprecipitated with H3K9me2 antibody, then analyzed by real-time PCR using primers for the RE-1 sites of NF-M, SYN1, SNAP25, and MAP-2 genes. *p, **p (vs. control), #p, ##p (vs. 100 μM H2O2-treated cells) (t-test, *p < 0.05, mean ± SD, n = 3).
![]()
Inhibition of G9a prevents neuronal cell damage caused by exposure to H2O2
To investigate the mechanism by which H2O2-damaged cells recover on inhibition of G9a, SH-SY5Y cells were incubated with or without BIX01294, prior to exposure to H2O2. The cells were then differentiated in neuronal induction medium. As shown in Fig. 4A, differentiation (3 days) declined in H2O2-treated cells and partially recovered on BIX01294 treatment; this was most evident at 7 days after differentiation (Fig. 4A). To confirm these results, immunoblot analyses were performed with neuron-specific antibodies to NF-M and MAP-2, which showed that expression levels of neuronal proteins decreased in H2O2-treated cells and increased on treatment with BIX01294 (Fig. 4B, C). Consistent results were obtained when used another G9a inhibitor UCN0321 (Fig. 5).

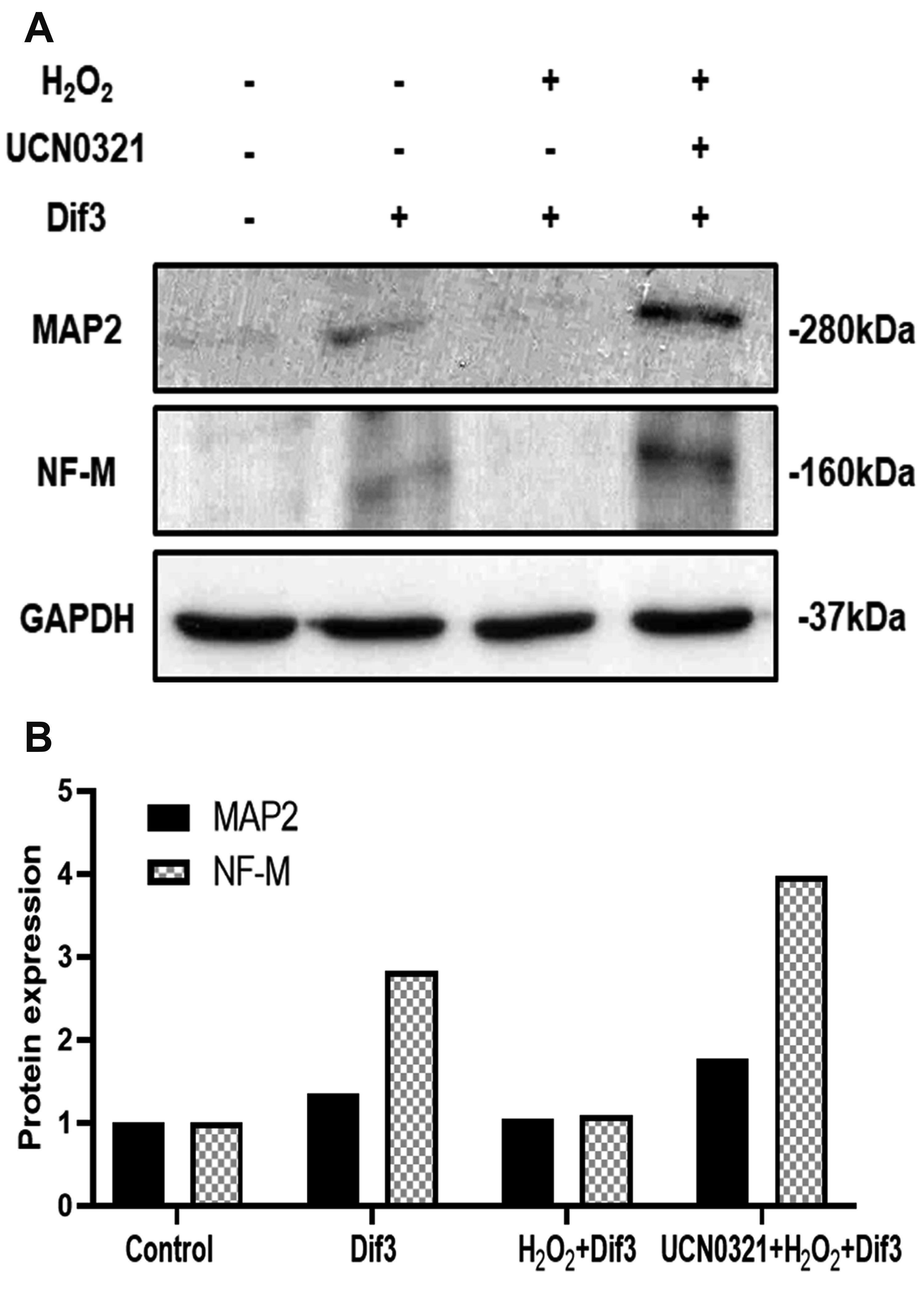
Fig. 4
Neuronal differentiation in 2O2-treated SH-SY5Y cells recovered on inhibition of G9a.
(A) SH-SY5Y cells were differentiated into neuron-like cells for 0–7 days. Each cell was visualized by light microscopy. (B, C) The expression levels of RE-1-containing neuron-specific proteins were examined using antibodies specific to NF-M and MAP-2 in H2O2-treated differentiated SH-SY5Y cells, with or without BIX01294 treatment. Ctrl indicates controls, which were non-differentiated SH-SY5Y cells. Sham indicates incubation with FBS 0.1% medium. β-actin was used as the internal standard.
![]()
Fig. 5
Recovery by another G9a inhibitor UCN0321 in H2O2-treated SH-SY5Y cells.
(A) SH-SY5Y cells were differentiated into neuronal cells for 3 days. The expression levels of NF-M and MAP-2 were measured in H2O2-treated cells, with or without UCN0321 treatment. GAPDH was used as the internal standard. (B) The expression levels were quantified using ImageJ software.
![]()
DISCUSSION
The human body contains a variety of specialized cell types with multiple functional roles. Although all human cells share the same genome, they differ in structure, morphology, function, and gene expression. Such differences are achieved through several means, including epigenetic modifications [7,8]. Epigenetic modifications are associated with both cellular differentiation and degeneration processes [24-26]. Our previous studies have shown that processes involved in the modulation of histone methylation, acetylation, and DNA methylation can promote neuronal differentiation and stimulate the expression of neuron-specific genes [22,27-29].
Histone methyltransferase G9a regulates the expression of RE-1-containing neuron-specific genes through binding to the REST complex [30]. Notably, G9a is activated by a variety of stimuli, including ethanol exposure and hypoxia, which can cause neurodegeneration [18,19,31]. A previous study showed that inhibition of G9a reverses β-amyloid-induced deficits in hippocampal neurons [32]. Thus, inhibiting aberrant activation of G9a is important for prevention of neurodegenerative diseases. In the present study, we investigated the relationship between neuronal cell death and aberrant activation of G9a due to oxidative stress exposure in SH-SY5Y neuroblastoma cells. As shown in Fig. 1B, cell viabilities decreased in H2O2-treated SH-SY5Y cells; such changes were partly accompanied by reduced levels of neuron-specific gene expression (Fig. 2).
Treatment with the G9a inhibitor BIX01294 prevented cell damage and restored the expression of neuron-specific genes (Fig. 1C–E and Fig. 2A, B). This indicated that the expression of neuronal genes may be disturbed by oxidative stress, thereby leading to neuronal cell death; moreover, G9a inhibition can attenuate these effects. Thus, the effects of oxidative damage are only partially mitigated on treatment with BIX01294; this may be a result of partial recovery of neuron-specific gene expression, and there may be a need for ROS scavenging by antioxidants to facilitate complete recovery.
The reduction of neuron-specific proteins is a critical element in the progression of neurodegeneration [33-35]. Sustained expression of neuron-specific genes may delay degeneration and attenuate neuronal cell death. The expression of neuron-specific marker genes is reduced by exposure to H2O2 for 12 h and recovers when treated with the G9a inhibitor BIX01294; notably, G9a activity was elevated in cells exposed to H2O2 (12 h). As shown in Fig. 3A, when SH-SY5Y cells were treated with 100 μM H2O2, H3K9me2 levels significantly increased. Furthermore, H3K9me2 levels in SH-SY5Y cells showed a weakly decreasing trend on treatment with BIX01294. This suggests that H3K9me2 may be moderately increased on exposure to oxidative stress, in the absence of G9a. The results of the ChIP assay showed that H3K9me2 was significantly increased in the proximal region of RE-1-containing neuron-specific genes in H2O2-treated cells, compared with that in the same region in control cells (Fig. 3B); however, H3K9me2 decreased in cells that were treated with both BIX01294 and H2O2. This suggests that oxidative stress causes activation of G9a, thereby interrupting the transcription of neuron-specific genes. Thus, aberrant expression of neuronal genes may interfere with the function of neuronal cells, and may lead to transformation and damage, ultimately resulting in cell death of varying severity. Treatment with BIX01294 may restore the expression of neuron-specific genes.
The activation of G9a blocks neuronal differentiation and leads to neurodegeneration; thus, we examined neuronal differentiation potency by using SH-SY5Y cells treated with or without BIX01294, prior to H2O2 exposure. The neuronal differentiation rate decreased in H2O2-exposed SH-SY5Y cells; this change was attenuated on inhibition of G9a (Fig. 4A, B). These results suggest that BIX01294 can restore the expression levels of neuron-specific proteins that were downregulated on oxidative stress-induced aberrant activation of G9a.
In the present study, we aimed to characterize the relationships among oxidative stress, G9a, and neurodegeneration. However, there were some limitations like, this study was limited to in vitro analysis, need to test with inhibition of G9a gene, and thus may not be entirely reproducible in vivo. Therefore, further studies are needed to confirm these findings regarding the relationships among aberrant activation of G9a, oxidative stress, and neurodegenerative diseases.
Oxidative stress induces aberrant activation of G9a, which leads to epigenetic modification of RE-1-containing neuron-specific genes; this disturbs neuronal gene expression and results in neuronal cell death, which is an important etiology of neurodegenerative diseases. Treatment with the G9a inhibitor BIX01294 can attenuate this aberrant G9a activity and protect against neuronal damage. Thus, the results of the present study suggest that G9a inhibition may provide a useful approach for treatment of neurodegenerative diseases caused by chronic exposure to oxidative stress.
SUPPLEMENTARY MATERIALS
Supplementary data including four tables can be found with this article online at https://doi.org/10.4196/kjpp.2021.25.1.51.
 XML Download
XML Download