

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Elevated blood pressure act as major risks of stroke, cardiovascular diseases, and kidney failure, which are leading causes of death globally [1], and it is known that increased salt consumption, caused by increased consumption of sodium-rich fast foods, induces serious health conditions [1].
Experimental studies showed that high-salt containing diets are related with hypertension development and induce cardiovascular diseases in hypertensive animals [2-4]. In normotensive rodent animals, high-salt diets have been reported to induce structural changes of arterioles, reduction of microvascular density, and impaired skeletal muscle vessel relaxation by vasodilatory stimuli [2,5-7].
It has not been established how salt induces hypertension, but many studies showed macrophage involvement in its pathogenesis. Circulating monocytes from hypertensive patients showed more pro-inflammatory phenotype than normotensive controls [8,9], and hypertensive patients showed higher levels of interleukin (IL)-2, IL-6, IL-8, IL-1β, IL-1α, IL-18, tumor necrosis factor-alpha (TNF-α), interferon gamma (IFN-γ), and monocyte chemoattractant protein-1, C-reactive protein (CRP) [10,11].
These inflammatory cytokines are involved in macrophage polarization, as defined by two phenotypes, that is, the macrophage type 1 (M1) and type 2 (M2) phenotype (pro- and anti-inflammatory phenotypes, respectively). The M1 phenotype induces pro-inflammatory mediators, such as, IL-6, TNF-α, and inducible nitric oxide synthase (iNOS) [12-14], whereas the M2 phenotype reduces responsiveness to IFN-γ which leads to the inductions of low levels of pro-inflammatory mediators and the upregulations of IL-10 and arginase-1 (Arg-1) [12,13,15]. Interestingly, one study demonstrated that macrophages which cultured in sodium-chloride enriched medium showed polarization toward M1 phenotype [16].
Gamma-aminobutyric acid (GABA) is a non-protein amino acid which acts as main inhibitory neurotransmitter in the central nervous system. Furthermore, the GABA has been reported to have hypotensive effects [17], for example, low-dose oral GABA taking has been reported to have a hypotensive effect in Wistar-Kyoto rats and spontaneously hypertensive rats [18]. The role played by GABA in cardiovascular diseases, such as, hypertension remains to be clarified, although it has been suggested it reduces the productions of some classical pro-inflammatory cytokines and regulates immune responses [18]. One study showed GABA-overexpressing white mice had lower morning systolic blood pressures (BPs) than control mice [19], which suggested GABA supplementation in food might reduce hypertension. Furthermore, several studies have demonstrated various food products containing GABA showed blood pressure decreasing effects in hypertensive patients [17,20-22]. GABA-salt is a fermented salt containing GABA prepared by fermentation with Lactobacillus brevis BJ20 as an alternative for the traditional salt, known as a cause of cardiovascular diseases, especially hypertension induced by sodium sensitivity [23,24]. GABA-salt and low sodium GABA-salt were previously prepared through the fermentation with Lactobacillus brevis [25,26]
In this study, we produced a GABA-salt by mixing the fermented solution containing GABA with refined salt and post-fermentation. We evaluated the effect of GABA-salt on hypertension in a high salt and high cholesterol diet (HCD)-induced mouse model of hypertension. We hypothesized GABA in the GABA-salt might reduce classical pro-inflammatory cytokines induced by M1 polarization and that this would attenuate hypertension.
METHODS
Preparation of GABA-salt
Lactobacillus brevis BJ20 was inoculated into a seed medium (3% yeast extract, 1% glucose, 1% monosodium glutamate, 95% water) previously sterilized at 121°C for 15 min by autoclaving. Ten percentage (v/v) seed medium previously cultured at 37°C for 24 h was inoculated into a sterilized main culture medium (2% yeast extract, 1% glucose, 6% L-glutamic acid, 91% water), fermented at 37°C for 24 h. Fermented solution was further fermented with 40% (w/w) refined salt for 6 h and then filtered (Advantec MFS Inc., Dublin, CA, USA) and spray dried to yield GABA-salt.
The refined salt (Hanju Corp., Yangju, Korea), which has been removed most of the impurities through filtration, precipitation and concentration processes using a large amount of seawater was purchased from a local market.
A DIONEX Ultimate 3000 HPLC system (Thermo Fisher Scientific, Waltham, MA, USA) equipped with a UV detector was used to confirm the presence and to determine the level of GABA in GABA-salt. The GABA-salt was diluted with distilled water to 1,000 mg/L. Gradient elution was performed with distilled water to 500, 250, 125, 62.5, 31.25, 15.625 ppm of standard solution. Four percentage of GABA-salt solution was diluted with distilled water then eluted 50 times with 0.02 N hydrochloric acid and passed through a membrane filter. The column was operated in descending mode at a flow rate of 1.0 ml/min and injection volume was 20 µl. Ultra-high-performance liquid chromatograph was performed using a two-phase solvent system comprised of 50 mM sodium acetate (pH 6.5, solution A) and acetonitrile/methanol/water (45:45:10 v/v/v, solution B). Zero minute, 90% of solution A and 10% of solution B and to 12 min, 60% of solution A and 40% of solution B and to 16 min, 10% of solution A and 90% of solution B were used. The column flow rate was maintained at 1.0 ml/min and injection volume was 20 µl. The GABA content (%) of GABA-salt is calculated by dividing the concentration (ppm) of GABA in the test solution by the total concentration (ppm) of the test solution after HPLC analysis and multiplying by 100.
Animals and GABA-salt administration
Male C57BL/6N mice (7-week old) were purchased from Orient Bio (Seongnam, Korea) and maintained under controlled conditions (23°C, 50% of humidity), and a 12/12 h dark/light cycle. The mice were fed either normal chow diet (ND) or a HCD (Harlan Laboratories, Indianapolis, IN, USA) for 5 weeks. Considering that GABA contains 0.76% of GABA-salt, mice were orally administered eating water with ND (ND group), salt with HCD (HCD/salt group; 0.03 mg/kg/day) or GABA-salt with HCD (HCD/GABA-salt group; 0.034 mg/kg/day) during the same 5 weeks.
After this 5-week feeding period, we measured water intake, urine volume and serum creatinine and blood urea nitrogen (BUN) and mice were sacrificed in accordance with guideline issued by the Institutional Animal Care and Use Committee of Gachon University (approval number: LCDI-2018-0066). In additional, we measured systolic, diastolic, and mean artery pressure at 3, 4, and 5 weeks. The BPs were monitored using a non-invasive tail-cuff CODA system (Kent Scientific Corp., Torrington, CT, USA) according to the manufacturer’s directions.
Serum creatinine and BUN validation
When mice were sacrificed, whole blood was collected and serum was seperated using serum-separating tubes (BD Vacutainer; Thermo Fisher Scientific). The serum creatinine and BUN were measured using by ELISA kit according to the manufacturer’s directions (Abcam, Cambridge, UK).
Cell culture and in vitro model
Murine endothelial cells (SVEC 4–10) was purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA). The cells were grown in Dulbecco's modified Eagle's medium (DMEM; Hyclone, Chicago, IL, USA) supplemented with 10% fetal bovine serum (FBS; Merck Millipore, OH, USA) and 1% penicillin streptomycin (Hyclone). Murine vascular smooth muscle cells (MOVAS) were also purchased from the ATCC and grown in DMEM (Hyclone) supplemented with 10% FBS (Merck Millipore) and antibiotics 0.2 mg/ml G-418 (Sigma-Aldrich, Burlington, MA, USA). Murine macrophages (Raw 264.7) were purchased from the Korea Cell Line Bank (Seoul, Korea). The cells were grown in DMEM supplemented with 10% FBS and 1% penicillin streptomycin.
The SVEC 4–10, MOVAS and Raw 264.7 cells were treated with 20 mM salt or 20 mM GABA-salt at the same concentrations used in the animal experiment. The salt and GABA-salt were dissolved in cell culture medium and stored at –4°C.
To make M1 macrophage activation model, 1M refined salt (Hanju Corp.) solution which solved in distilled water is prepared. The salt solution was mixed and diluted with the media up to 20 mM concentration. Then the cell was incubated in culture plate for 4 days and harvested for experiments.
RNA extraction and cDNA synthesis
Total RNA in aorta tissue and cells was isolated using RNAiso Plus kits (TAKARA, Tokyo, Japan) [27]. Animal samples and cell samples are mixed with 0.2 ml of chloroform (Amresco, Solon, OH, USA) and then, centrifuged at 13,000 ×g for 15 min at 4°C. Clear part was collected into new tubes and mixed with 0.4 ml of 100% isopropanol and centrifuged at 13,000 ×g for 10 min at 4°C. Isolated RNA pellets were washed with 1 ml of 75% ethanol and centrifuged at 7,500 ×g for 5 min at 4°C. Dried pellets were dissolved in 10 to 50 µl diethyl pyrocarbonate (DEPC, Sigma-Aldrich) treated water and the prepared RNA was quantified using a Nanodrop 2000 (Thermo Fisher Scientific). Complementary DNA (cDNA) was synthesized from RNA using a cDNA synthesis kit (PrimeScript; TAKARA).
Quantitative real time polymerase chain reaction (qRT-PCR)
qRT-PCR was performed to determine mRNA levels. Briefly, primer and distilled water were first mixed and placed in 384-well plate. Template cDNA and SYBR green reagent (TAKARA) were then added to the well plate and spun down (Eppendorf, Hamburg, Germany). Mixed samples were validated using by qRT-PCR machine (Bio-Rad, Hercules, CA, USA). All genes of interest are listed in Supplementary Table 1.
Immunofluorescence for aortic tissues
Aorta paraffin block tissues were sectioned at 7 um and placed on slides, dried at 40°C for 24 h, de-paraffinized, and incubated in normal animal serum containing 0.03% triton X-100 to block antibody binding. Slides were then incubated with antibodies (listed in Supplementary Table 2) for 2 days at 4°C, rinsed 3 times with phosphate-buffered saline (PBS), incubated for 1 h at room temperature with fluorescence conjugated secondary antibodies, and rinsed 3 times with PBS. They were then incubated with 4´6-diamidino-2-phenylindole solution (DAPI; Sigma-Aldrich) at room temperature for 5 min, rinsed 3 times with PBS and mounted with cover slips using vector shield solution (Vector Laboratories, Burlingame, CA, USA). The fluorescent signal was detected using a confocal microscope (LSM 710; Carl Zeiss, Oberkochen, Germany).
Immunofluorescence for cells
SVEC 4–10 and MOVAS cells (104) were cultured in an 8-well chamber (Thermo Fisher Scientific) for 24 h, removed from medium, and rinsed 3 times with PBS. They were then fixed in cold 100% methanol (Duksan Company, Ansan, Korea) at room temperature for 15 min, rinsed 3 times with PBS, incubated in normal animal serum containing 0.03% triton X-100 to block antibody binding, incubated with antibodies (listed in Supplementary Table 2) at 4°C for 1 day, and then rinsed 3 times with PBS. The cells were then incubated for 1 h at room temperature with fluorescence conjugated secondary antibodies, rinsed 3 times with PBS, incubated for 5 min at room temperature in DAPI solution, and rinsed 3 times with PBS. Finally, the cells were mounted with cover slips using vector shield solution. The fluorescent signal was detected using a confocal microscope.
Immunohistochemistry using 3, 3´-diaminobenzidine
Aorta paraffin block slides were de-paraffinized and incubated with 0.3% H2O2 (Sigma-Aldrich) for 30 mins, rinsed 3 times with PBS, incubated in normal animal serum to block antibody binding, incubated with antibodies (listed in Supplementary Table 2) for 2 days at 4°C and then rinsed 3 times with PBS. Slides were then treated with biotinylated secondary antibodies in the ABC kit (Vector Laboratories), incubated for 1 h at room temperature in blocking solution, and rinsed 3 times with PBS. Slides were developed with 3, 3´-diaminobenzidine (DAB) substrate containing 0.3% H2O2 at room temperature for 15 min and mounted with cover slips using DPX mounting solution (Sigma-Aldrich). Stained images were visualized by light microscopy (Olympus Optical Co., Tokyo, Japan) [28].
Terminal deoxyribonucleotidyl transferase-mediated biotin-16-dUTP nick-end labelling assay
To count numbers of apoptotic endothelial cells on paraffin block slides of aortas, we used Terminal deoxyribonucleotidyl transferase (TdT)-mediated biotin-16-dUTP nick-end labelling (TUNEL) assay kit (TransGen Biotech Co., Ltd., Beijing, China). The slides were stained with DAPI solution (Sigma-Aldrich) at room temperature for 5 min, washed 3 times with PBS, and mounted with cover slips using vector shield solution (Vector Laboratories). The fluorescence was detected using a confocal microscope (LSM 710).
Histological H&E staining
Paraffin block slides of aortas were de-paraffinized, incubated in hematoxylin (DAKO, Carpinteria, CA, USA) for 1 min, rinsed with water for 3 min at room temperature, incubated with eosin (Sigma-Aldrich) for 15 sec, rinsed with water, and dehydrated using an alcohol gradient. Finally, slides were mounted with cover slips using DPX mounting solution and visualized by light microscopy (Olympus Optical Co.) [29]. Intima-media thickness ratios were measured using the Image J program (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Results are presented as means ± standard deviation (SD). The significances of intergroup differences were determined using the Mann–Whitney U-test for multiple comparison and the Kruskal-Wallis test for post-hoc. The statistical analysis was conducted using SPSS 22 statistical software (IBM Co., Armonk, NY, USA), and statistical significance was accepted for p-values ≤ 0.05.
RESULTS
Composition of GABA-salt
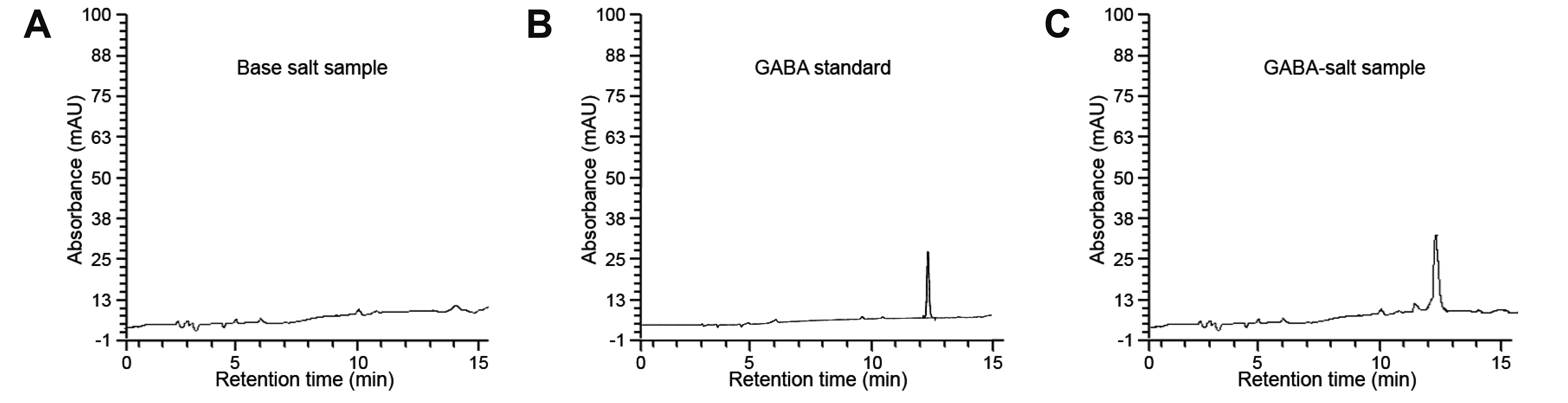
GABA-salt attenuated BP increases and renal function deterioration in hypertensive mice
Mean systolic BPs in the HCD/salt fed animals were significantly higher than in the ND controls at all time points during the 5-week experimental period. Mean systolic BP in HCD/GABA-salt fed animals was significantly lower than in HCD/salt fed animals at all time points. Mean systolic BPs of HCD/GABA-salt fed animals were higher than those of ND controls, but not significantly so (Fig. 2A). Mean diastolic BPs in HCD/salt fed animals were significantly higher than in ND controls at all time points. Mean diastolic BPs in HCD/GABA-salt fed animals were significantly lower than in HCD/salt fed animals at 4 and 5 weeks. Mean diastolic BPs in HCD/GABA-salt fed animals were higher than in ND controls, but not significantly so at any time point (Fig. 2B). Mean arterial pressures (MAPs) in HCD/salt fed animals were significantly higher than in ND controls at all time points. MAPs increase observed in HCD/salt fed animals were attenuated in GABA-salt fed animals (Fig. 2C). In additional, water intake (Fig. 2D) and urine volume (Fig. 2E) were in HCD/salt fed animals were significantly higher than in ND controls. The water intake and urine volume in HCD/GABA-salt fed animals were significantly lower than in HCD/salt fed animals at 5 weeks. In hypothalamus, primary osmoreceptors modulate arginine vasopressin (AVP) which regulates the tonicity of systemic fluids. The AVP mRNA level was in HCD/salt fed animals were significantly higher than in ND controls. The AVP level in HCD/GABA-salt fed animals was significantly lower than in HCD/salt fed animals at 5 weeks (Supplementary Fig. 1).

Fig. 2
Regulation of blood pressure, water intake, urine volume and renal function of gamma-aminobutyric acid (GABA)-salt in high cholesterol (HCD) diet-induced mouse model.
Blood pressures were measured at 3, 4 and 5 weeks. (A–C) Graphs of the blood pressures of mice fed a normal diet (ND, circle), a high cholesterol diet and normal saline (HCD/salt, square) diet, or a high cholesterol and GABA-salt (HCD/GABA-salt, triangle) diet showing; (A) systolic blood pressures, (B) diastolic blood pressures, and (C) mean arterial blood pressures. (D) Water intake and (E) urine volume were measured at 5 weeks and blood chemistry tests including (F) serum creatinine and (G) BUN show kidney function in ND group, HCD/salt group and HCD/GABA-salt group. BUN, blood urea nitrogen. *p < 0.05, **p < 0.01, and ***p < 0.001 vs. ND; and $p < 0.05, $$p < 0.01, and $$$p < 0.001 vs. HCD/Salt.
![]()
Interestingly, renal function test including creatinine (Fig. 2F) and BUN (Fig. 2G) serum level were in HCD/salt fed animals were significantly higher than in ND controls. The water intake and urine volume in HCD/GABA-salt fed animals were significantly lower than in HCD/salt fed animals at 5 weeks. Body weight among three groups was not statistically significant. Total cholesterol was statistically significant in HCD/salt group but decreased in HCD/GABA-salt group (Supplementary Fig. 2).
GABA-salt attenuated M1 polarization and cytokine secretion in the aortic tissues of hypertensive mice and in Raw 264.7 macrophage
Results of immunohistochemistry and qRT-PCR showed CD86 expression was significantly higher in HCD/salt fed animals than in ND controls, and that these increases were significantly less in HCD/GABA-salt fed animals (Fig. 3A, B and Supplementary Fig. 3). The expression of TNF-α (an M1 inflammatory cytokine) was higher in those of HCD/salt fed animals than in ND controls, but was significantly lower in HCD/GABA-salt fed animals than in HCD/salt fed animals, as determined by qRT-PCR and immunohistochemistry. iNOS expressed by M1 macrophages levels were higher in HCD/salt fed animals than in ND controls, and lower in HCD/GABA-fed animals than in HCD/salt fed animals (Fig. 3C). CD206 expression was significantly higher in HCD/salt fed animals than in ND controls. CD206 expression in HCD/GABA-salt fed animals was non-significantly lower than HCD/salt fed animals (Fig. 3D, E and Supplementary Fig. 3). Transforming growth factor beta 1 (TGF-β) secreted by M2 macrophages levels were lower in HCD/salt fed animals than in controls, but significantly higher in HCD/GABA-salt fed animals than in HCD/salt fed animals. Arg-1 levels expressed by M2 macrophages were lower in HCD/salt fed animals than in ND controls, but, Arg-1 level in HCD/GABA-salt fed animals was non-significantly higher than HCD/salt fed animals (Fig. 3F).

Fig. 3
Inhibitory effects of gamma-aminobutyric acid (GABA)-salt on expressions of type 1/type 2 macrophage (M1/M2) and associated cytokines in high cholesterol diet (HCD)-induced mouse model.
(A) Immunohistochemistry images showing M1 macrophages (CD86; brown, arrows). Scale bar = 40 µm. (B) The qRT-PCR graph showing mRNA expressions of M1 (CD86) macrophage marker. (C) The qRT-PCR graph showing the mRNA expressions of M1 related cytokines including TNF-α and iNOS. (D) Immunohistochemistry images showing M2 macrophages (CD206; brown, arrow). Scale bar = 40 µm. (E) The qRT-PCR graph showing mRNA expressions of M2 (CD206) macrophage marker. (F) The qRT-PCR graph showing the mRNA expressions of M2 related cytokines including TGF-β and Arg-1. NS, not significant compared to HCD/salt. *p < 0.05, and **p < 0.01 vs. normal diet (ND) and $p < 0.05, and $$p < 0.01 vs. HCD/salt.
![]()
We also treated Raw 264.7 macrophages with salt or GABA-salt (Fig. 4). CD86 mRNA levels in salt treated macrophages were significantly higher than in media treated macrophages, but lower in GABA-salt treated macrophages than in salt treated macrophages (Fig. 4A). CD206 expression reduced when macrophages were treated with salt but were higher in GABA-salt treated macrophages than in salt treated macrophages (Fig. 4B). TNF-α expression in salt treated macrophages was significantly higher than in media treated macrophages, but lower in GABA-salt treated macrophages than in salt treated macrophages (Fig. 4C). TGF-β expression in salt treated macrophages was significantly lower than in media treated macrophages, but higher in GABA-salt treated macrophages than in salt treated macrophages (Fig. 4D).

Fig. 4
Inhibitory effects of gamma-aminobutyric acid (GABA)-salt on the expressions of type 1 macrophages (M1) and associated cytokines in vitro.
(A, B) qRT-PCR graph showing the mRNA expressions of M1 and M2 macrophage marker in media, salt or GABA-salt treated Raw 264.7 cells. (C, D) qRT-PCR graph showing the mRNA expressions of M1 (TNF-α) and M2 (TGF-β) macrophage marker in media, salt or GABA-salt treated Raw 264.7 cells. (E, F) qRT-PCR graph showing the mRNA expressions of M1 (iNOS) and M2 (Arg-1) macrophage marker in media, salt or GABA-salt treated Raw 264.7 cells. NS, not significant compared to salt. *p < 0.05 vs. media; $p < 0.05 vs. salt.
![]()
The iNOS expression was higher in salt treated macrophages than in media treated macrophages, and lower in GABA-salt treated macrophages than in salt treated macrophages (Fig. 4E). On the other hand, Arg-1 expression was lower in salt treated macrophages than in the media treated macrophages and substantially higher in GABA-salt treated macrophages than in salt treated macrophages (Fig. 4F).
GABA-salt preserved expressions of GABAB receptor, eNOS and of phosphorylated eNOS in endothelial cells and endothelial cell dysfunction and cell death in the aortas of hypertensive animals
GABAB receptor expression levels were lower in salt treated endothelial cell (ECs) than in media treated ECs, and GABA-salt attenuated these reductions in GABAB receptor expression (Fig. 5A). Immunohistochemistry results revealed that eNOS and phosphorylated eNOS levels were lower in salt treated ECs than in media treated ECs, and that their levels were higher in GABA-salt treated ECs than in salt treated ECs (Fig. 5B and Supplementary Fig. 4A). eNOS and phosphorylated eNOS levels were assessed immunohistochemically in aortic EC layers. The levels of both proteins were lower in HCD/salt fed animals than in ND controls, and levels were higher in HCD/GABA-salt fed animals than in HCD/salt fed animals (Fig. 5C and Supplementary Fig. 4B).

Fig. 5
Effects of gamma-aminobutyric acid (GABA)-salt on the expression and function of GABAB receptor in endothelial cell (ECs).
To identify changes in GABAB receptors and in peNOS and eNOS, media, salt or GABA-salt treated ECs and aorta of hypertensive mice were subjected to qRT-PCR and immunofluorescence. (A) The qRT-PCR graph showing the mRNA expressions of GABAB receptor in media, salt or GABA-salt treated SVEC 4–10. (B, C) Immunofluorescent images showing eNOS and peNOS (green, nuclei; blue) expressions in (B) SVEC 4–10 and in (C) the aorta of hypertensive mice (brown, arrows). Scale bar = 50 µm. (D) Immunofluorescent images showing the expression of the adherent proteins such as E-selectin, VCAM-1, ICAM-1 and vWF (green, nuclei; blue) in the aorta of hypertensive mice. Scale bar = 50 µm. (E, F) Apoptotic cells are detected by TUNEL analysis. The fluorescent images and graph showing apoptotic cell in the aorta of hypertensive mice. Scale bar = 50 µm. ICAM-1, Intercellular Adhesion Molecule-1; VCAM-1, vascular cell adhesion molecule-1; vWF, von Willebrand Factor. *p < 0.05, and ***p < 0.001 vs. culture media or normal diet (ND); and $p < 0.05, and $$$p < 0.001 vs. salt or high cholesterol diet (HCD)/salt.
![]()
Protein levels of adhesion molecules, that is, E-selectin, Intercellular Adhesion Molecule-1 (ICAM-1), vascular cell adhesion molecule-1 (VCAM-1), and von Willebrand Factor (vWF) were higher in endothelial layers in the HCD/salt fed animals than in ND controls, and lower in HCD/GABA-salt than in HCD/salt fed animals (Fig. 5D and Supplementary Fig. 4C). Endothelial cell death was greater in HCD/salt fed animals than in ND controls, and was lower in HCD/GABA-salt than in HCD/salt fed animals (Fig. 5E, F).
GABA-salt preserved GABAB receptor expression and attenuated Endothelin-1 expression in VSMCs and SMC proliferation and medial thickening in the aortas of hypertensive animals
GABAB receptor expression levels in salt treated VSMCs and ECs were lower than in their media treated counterparts, and higher in GABA-salt treated than in salt treated VSMCs and ECs. GABAB receptor expression was lower in salt treated ECs and VSMCs than in controls, but higher in GABA-salt treated ECs and VSMCs than in salt treated counterparts (Figs. 5A, 6A).

Fig. 6
Effects of gamma-aminobutyric acid (GABA)-salt on the expression of GABAB receptor and its function in vascular smooth muscle cells (VSMCs).
(A) The qRT-PCR graph showing the mRNA expressions of GABAB receptor in media, salt or GABA-salt treated mouse vascular smooth muscle cell (MOVAS). (B) Immunofluorescent images showing Endothelin-1 (ET-1) (green, nuclei; blue) expressions in media, salt or GABA-salt treated MOVAS. Scale bar = 50 µm. (C, D) Immunohistochemistry staining image and graph showing proliferating cell nuclear antigen (PCNA), as proliferating cell marker, positive cell (brown, arrows) in hypertensive mice. Scale bar = 50 µm. H&E staining image and graph showing changes of vessel thicknesses in hypertensive mice. Scale bar = 500 µm. *p < 0.05, and **p < 0.01 vs. culture media or normal diet (ND); $p < 0.05 vs. salt or high cholesterol diet (HCD)/salt.
![]()
Immunohistochemistry results showed that Endothelin-1 (ET-1) expression levels in salt treated VSMCs were greater than in media treated VSMCs, and lower in GABA-salt treated VSMCs than in salt treated VSMCs (Fig. 6B and Supplementary Fig. 5). In this study, the ET-1 level was higher in salt treated VSMCs than in media treated VSMCs and lower in the GABA-salt treated VSMCs than salt treated VSMCs (Fig. 6B and Supplementary Fig. 5). Furthermore, SMC proliferation and medial thickening were attenuated in HCD/GABA-salt fed animals (Fig. 6C, D). Proliferating cell nuclear antigen (PCNA) staining was used to evaluate SMC proliferation in medial aortic layers. The PCNA expression in HCD/salt fed animals was higher than in ND controls, but lower in HCD/GABA-salt than in HCD/salt fed animals. In addition, intima-media ratios were higher in HCD/salt fed animals than in ND controls, and lower in HCD/GABA-salt than in HCD/salt fed animals (Fig. 6C, D). In the present study, PCNA staining showed SMC proliferation was greater in HCD/salt fed animals and that intima-media ratios were higher in the HCD/salt fed animals than in ND controls (Fig. 6C, D).
DISCUSSION
Several animal studies have demonstrated that consumption of high salt induces hypertension in rats and primates [30,31]. Similarly, BPs were increased in mice by HCD and salt in our study. It has been reported the risks of developing a high BP are increased by 14% and 27% when daily salt intakes are 12 or 18 g/day, respectively, as compared with a daily salt intake of 6 g/day [32,33]. It is difficult for Asians to reduce salt intake, because Asians put more salt or condiments containing salt such as soy sauce during cooking than Caucasians [32,33]. Thus, we believe that GABA-salt might provide those who find it difficult to reduce salt intake as a means of reducing BPs. Although the mechanism responsible for salt-induced hypertension has not been fully elucidated, several studies have shown NaCl stimuluses the secretions of TNF-α [34,35]. TNF-α is a typical M1 macrophage related cytokine and is secreted mainly by macrophages [36]. Thus, it seems that M1 macrophages or the cytokines they secrete might have a primary role in the hypertension induced by salt intake. In this study, CD 86 (a M1 macrophage marker) level and TNF-α and iNOS (expressed by M1 macrophages) levels were elevated in HCD/salt fed animals (Fig. 3). Furthermore, similar results were obtained from our in vitro studies, that are, salt treated macrophages polarized toward the M1 type and expressed more TNF-α and iNOS than media treated macrophages (Fig. 4). It has been reported iNOS expressed by M1 macrophages promote endothelial dysfunction [37], and that hypertension is related with EC dysfunctions lead to decreasing endothelial-dependent vasodilation and damage [38-42].
The pathophysiology of hypertension is not fully revealed, but several studies have shown that the inhibition of M1 macrophage activation causes BP normalization, which suggests the M1 phenotype is a key factor of hypertension [11,43,44]. In this study, TNF-α and iNOS were found to be over-expressed in hypertensive mice (in vivo, HCD/salt group) and in salt treated macrophages (in vitro, salt group), and these over-expressions of TNF-α and iNOS were attenuated by feeding animals HCD/GABA-salt (in vivo, HCD/GABA-salt group) and by treating macrophages with GABA-salt (in vitro, GABA-salt group).
In resting ECs, eNOS is primarily placed at cell membranes and in cytoplasm, and translocate close to the nucleus by agonists [45,46]. eNOS translocation leads to increase level of [Ca2+]i in ECs [47]. It has been shown GABAB receptors which expressed in cultured human arterial ECs regulate the translocations of eNOS and level of [Ca2+]i [47]. Furthermore, another study showed baclofen, agonist GABAB receptor, induced vasodilation of 50% of rat retina vessels, and that this vasodilation is inhibited by 2-hydroxysaclofen, the GABAB receptor antagonist [48].
Endothelial dysfunction leads to the increased expression levels of cell adhesion molecule (E-selectin, ICAM-1, and VCAM-1) [49], and it is known TNF-α enhanced the expressions of the adhesion molecules [50,51]. The E-selectin, ICAM-1, and VCAM-1 lead to leukocyte adhesion at inflamed sites [52], and these adhesion molecules lead to adhesion between leukocytes and ECs and transmigration into vessel walls [53]. In this study, E-selectin, ICAM-1, and VCAM-1 protein levels in aortic ECs were elevated in hypertensive animals. Furthermore, vWF expression was also elevated in hypertensive animals. vWF release is increased when ECs are damaged, thus, vWF is an indicator of endothelial dysfunction [54]. When eNOS is activated in ECs, it generates nitric oxide (NO) which leads to blood vessel dilation [55]. On the other hand, endothelial dysfunction reduces endothelium-dependent vasodilatory response and eNOS availability [56]. Phosphorylation at serine residue 1179 of eNOS increases enzymatic activity which lead to NO production [57], whereas TNF-α compromises endothelium-dependent vasodilation by reducing eNOS synthesis and activity, and thus, decreasing the bioavailability of NO [57]. In this study, the expressions of eNOS and phosphorylated eNOS were found to be lower in hypertensive mice.
EC apoptosis relates with SMC migration and proliferation, increase blood coagulation, increase leukocyte infiltration, and cause endothelial dysfunction [58]. In this study, EC death and endothelial dysfunction were greater in hypertensive animals. Furthermore, the factors that induce endothelial dysfunction, such as, adhesion molecule (E-selectin, ICAM-1, and VCAM-1) and vWF levels, and EC death were lower in HCD/GABA-salt that in HCD/salt fed animals. In addition, the protein levels of eNOS and phosphorylated eNOS, which have a protective effect on EC damage, were higher in HCD/GABA-salt than in HCD/salt fed animals.
VSMCs phenotype could changes from the contractile phenotype to the synthetic phenotype by hypertensive stress [59]. In addition, it is known that vascular wall is thickened by activated VSMCs which are accumulated in intima and media [60]. TNF-α induced the expression of cell adhesion molecules, such as VCAM-1, and lead to vascular inflammation, which enhance vascular fibrosis and SMC proliferation and subsequently increased arterial stiffness [59].
These results indicate GABA-salt reduces M1 polarization and the expression levels of M1 macrophage related factors (iNOS and TNF-α) as compared with salt. The iNOS and TNF-α are major triggering factors of endothelial dysfunction and VSMC proliferation, which are known to induce hypertension. Although our results did not allow access to mechanism responsible for the protective effect of GABA on EC function, they do suggest GABAB receptor expression attenuation by GABA increases eNOS activity and protects EC from salt induced injuries. It is also possible that GABA reduced salt-induced endothelial dysfunction by controlling TNF-α induced monocyte adhesion, as it has been reported an increase endogenous levels of GABA were associated with decrease in monocyte adhesion [60].
Taken together, our findings suggest GABA might affect eNOS function and modulate macrophage adhesion to ECs at high salt consumption levels by reducing TNF-α expression, and thus, reducing the expressions of adhesion molecules and NOS inhibition in ECs, and by possibly reducing the expression of ET-1, which participates in vasoconstriction and reduces VSMC proliferation, and thus, causes luminal narrowing or vascular stiffness. Finally, and perhaps the most important observation is that GABA-salt fed animals had significantly lower BP increases than salt fed animals. GABA-salt caused lower BP increases in hypertensive mice by modulating endothelial cell dysfunction and VSMC proliferation.
SUPPLEMENTARY MATERIALS
Supplementary data including five figures and two tables can be found with this article online at https://doi.org/10.4196/kjpp.2021.25.1.27.
 XML Download
XML Download