

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
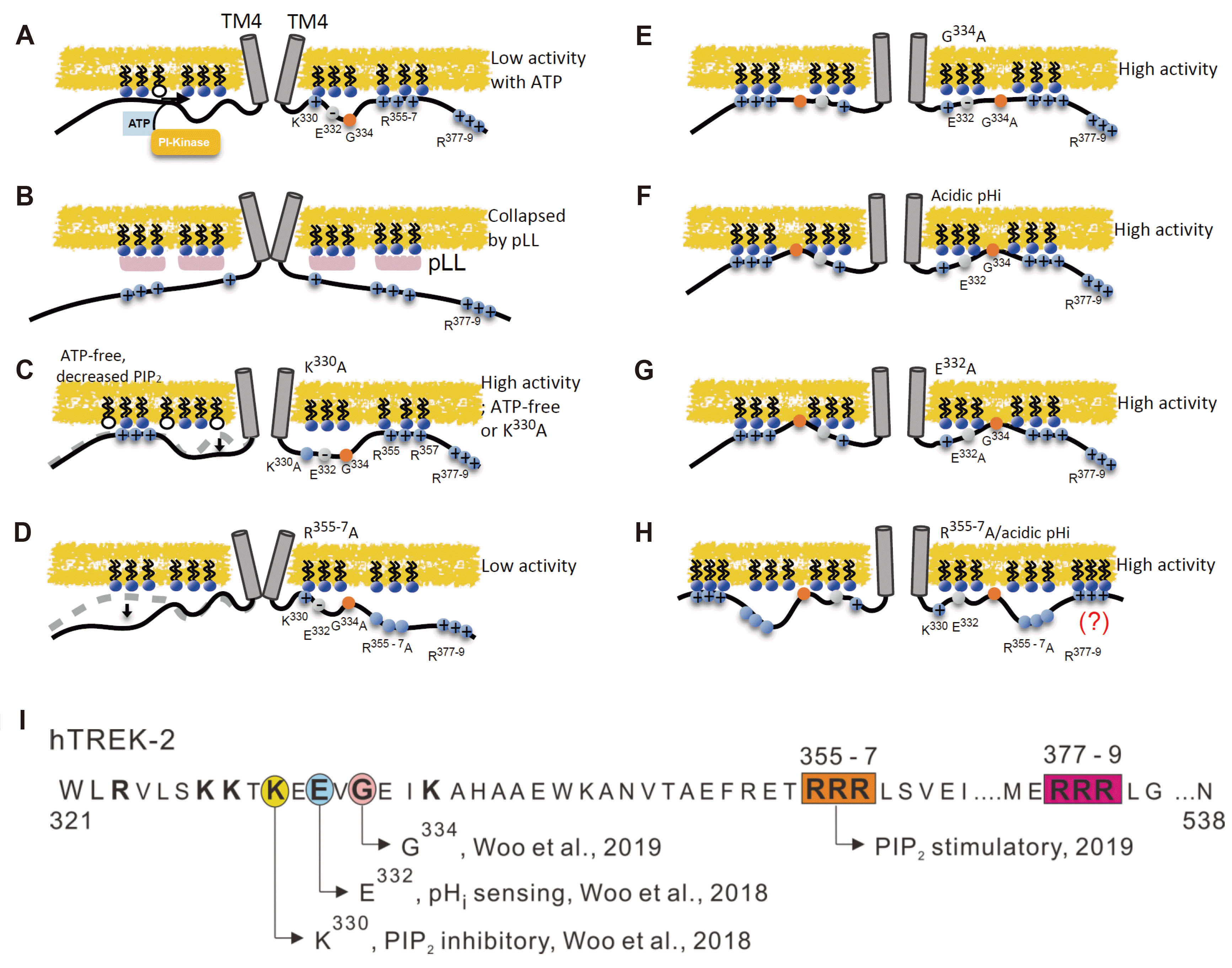
Two-pore domain K+ (K2P) channel family proteins coded by KCNK genes assemble as dimers to form a functional K+ channel. Each subunit has four transmembrane domains (TM1–TM4), two-pore domains (P1 and P2), and intracellular N- and C-termini. The K2P channels show voltage-independent activity, consistent with the “leak-type” background K+ conductance observed in various types of cells [1-3]. However, some members of the K2P channels show wide ranges of regulation by cellular signals and by various conditions. Especially, TWIK-related K+ channels (TREKs) such as TREK-1 (KCNK2) and TREK-2 (KCNK10) show sensitivity to physicochemical stimuli; activation by membrane stretch, polyunsaturated fatty acid, and acidic intracellular pH (pHi) [4-7]. Different from the other members of K2P channels, the TREKs show relatively low basal activity that is largely increased by the various kinds of stimuli. As for the low basal activity, tonic inhibition by the physiological level of phosphatidylinositol 4,5-bisphosphate (PIP2) has been suggested as the key mechanism including our previous studies (Fig. 1A) [8-14].
In general, basic amino acids such as Arg (R) and Lys (K) in the cytoplasmic domain of ion channels are responsible for the PIP2-dependent regulation via electrostatic binding with the anionic head groups of the phospholipids. The direction of regulation, activation or inhibition, appears controversial and diverse depending on the channels. Regarding the TREKs, their complete inhibition by cytoplasmic-side application of the polycationic PIP2 scavenger (e.g., poly-L-lysine [pLL]) initially indicated a simple stimulatory role of PIP2 (Fig. 1B) [9]. However, further investigations by us have shown more complex regulatory features; the inhibition by a relatively high level of PIP2 and the activation by a partial decrease of PIP2. The latter type of activation is demonstrated by the removal of cytoplasmic ATP that results in the spontaneous decrease of PIP2 by lipid phosphatase. Vice versa, the tonic inhibition by the physiological level of PIP2 maintained by the membrane-associated phosphoinositide (PI)-kinase and ATP [13] (Fig. 1A).
The dual-mode of TREK channel regulation by PIP2 appears to be concentration-dependent, and the site-directed mutagenesis studies of the proximal cytoplasmic C-terminal region (pCt) could elucidate the corresponding sites [13-15]. In terms of the human TREK-2 (hTREK-2) sequence, the Lys (K330) in the early pCt is responsible for the inhibitory regulation by a relatively higher level of PIP2 (Fig. 1C) [14]. In contrast, three consecutive Arg residues (R355-7) mediate the positive regulation by PIP2 (Fig. 1D) [15]. Although there are additional triple Arg residues (R377-9) distal to the R355-7, their mutation by Ala substitution (R377-9A) did not affect the activity of TREK-2 [15]. Therefore, it was assumed that the more distal R377-9 residues are not contributing to the regulation by PIP2, or might not be bound to the negatively charged phospholipids (Fig. 1A).
In addition to the mutually antagonizing roles of K330 and R355-7, an interesting role of Gly (G334) between them was suggested from our recent study; its mutation by Ala substitution (G334A) converts into a tonic active state, similar to the K330A (Fig. 1E) [15]. Because Gly could confer flexibility or bent conformation to the protein structure, the drastic changes of channel activity in G334A might reflect the outcome of a ‘tug of war’ between K330 and R355-7, i.e., dominating influence of the stimulatory R355-7 and putative loss of the electrostatic binding between K330 and PIP2 in G334A. However, such speculation could not exclude the possibility that the G334 mutation might have changed the channel property independent of the mutual interaction between K330 and R355-7.
Another key feature of TREK-2 is the activation by acidic pHi that is mediated by the titration of the negatively charged Glu (E332) close to the inhibitory K330 [10,11,13,14]. Our hypothetical model of the pHi-dependence is that the inhibitory influence from K330 could be relieved by the neutralization of E332 (Fig. 1F, G). In fact, the tonic high activity in K330A was not further activated by acidic pHi [14]. In the activation by acidic pHi, the putatively bent conformation at the nearly Gly residue (G334) appears critical because its mutation (G334A) also abolished the activation by acidic pHi as well as inducing tonic high activity (Fig. 1E) [15]. An interesting finding in our previous study was that the activation by acidic pHi could be still induced in R377-9A [15]. The result suggested a possibility that R377-9 might play a hidden role as the putative assistant binding with PIP2 (Fig. 1H), which was not investigated.
With this background, in the inside-out patch-clamp studies of the hTREK-2 and its various mutants, we compared their sensitivity to ATP and acidic pHi to get the clues to further understand the structure-function relation of TREK-2. The specific question was whether the combined mutation of G334 with K330 (K330A/G334A) or with R355-7 (G334A/R355-7A) show similar or different properties with the previous findings in K330A, G334A, and R355-7A. We hypothesized that if the putative bent conformation in the G334 of wild-type (WT) hTREK-2 simply mediates the tug-of-war between K330 and R355-7, the properties of double mutants, i.e., K330A/G334A and G334A/R355-7A, would be the same with the properties of K330A and R355-7A, respectively.
Apart from this question, we also asked whether the combined triple mutations of R355-7 and R377-9 with G334 (G334A/R355-7A/R377-9A) show similar or different properties with double mutants in G334 and R355-7 (G334A/R355-7A). The experimental result might elucidate a hypothetical hidden role of R377-9 in hTREK-2.
METHODS
Cell culture and preparation
Human embryonic kidney (HEK293T) cells were purchased from ATCC (Manassas, VA, USA) and incubated in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Grand Island, NY, USA) supplemented with 10% (50/500 ml) fetal bovine serum (FBS; Hyclone, Logan, UT, USA) and 1% penicillin-streptomycin (Gibco) at 37°C in 20% O2– 5% CO2.
Heterologous expression of hTREK-2
The human complementary DNAs (cDNAs) of TREK-2 (hTREK-2, SC222775) was purchased from ORIGENE and was transfected into HEK293T cells using a TurboFect transfection reagent (ThermoFisher Scientific, Waltham, MA, USA). Point mutations in hTREK-2 were generated using the QuickChange II Site-Directed Mutagenesis Kit (Agilent Technologies, Palo Alto, CA, USA). Sequences of the mutants were confirmed by DNA sequencing.
Electrophysiology
Cells were transferred into a bath mounted on the stage of an inverted microscope (TE2000-S; Nikon, Tokyo, Japan). The bath (0.15 ml) was perfused at 5 ml/min, and voltage-clamp experiments were performed at room temperature (22°C–25°C). Patch pipettes with a free-tip resistance of about 3–4 MΩ were used for inside-out (i-o) patch-clamp. The pipettes were connected to the head stage of a patch-clamp amplifier (Axopatch-1C; Axon Instruments, Foster City, CA, USA). pCLAMP software v.10.6 and Digidata-1322A (Axon Instruments) were used to acquire data and apply command pulses. The recorded currents were sampled at 10 kHz and were low-pass filtered at 2 kHz (i-o). The stored currents were analyzed using Clampfit v. 10.6 and Origin v.8.0 (Microcal, Northampton, MA, USA).
Solutions and chemicals
Both pipette solution and bath solutions of i-o patch-clamp contained (in mM) 145 KCl, 0.1 EGTA, 10 HEPES, and 20 sucrose with a pH of 7.0 (titrated with KOH). For acidic (pH 6.0, pH 5.0) solution applied to the cytoplasmic side of i-o patch-clamp, half of HEPES was substituted with 5 mM MES (2-[N-morpholino] ethanesulfonic acid). The chemicals and drugs used in this study were purchased from Sigma-Aldrich (St. Louis, MO, USA).
Statistical analysis
Data are represented as mean ± standard error of the mean (SEM). Results were compared using paired or unpaired two-sample t-test was employed for comparison of data collected from mutations. p-values < 0.05 were considered statistically significant. In the figures, *, **, and *** indicate p < 0.05, 0.01 and 0.001, respectively. All statistical analyses were done with Origin 2016 software (OriginLab, Northampton, MA, USA).
RESULTS
After making inside-out configuration of patch-clamp experiment, inward K+ current was recorded at –60 mV of holding voltage with symmetrical 145 mM [K+]. Since the activity of hTREK in the control state showed a tendency of spontaneous increase in the initial period after the membrane excision, we compared the responses to ATP and acidic pHi at 2–3 min confirming a steady-state.
Representative traces of the inward K+ currents from WT and the various mutants are shown in Fig. 2. The experimental protocols were, (1) changing pHi from 7.0 to 6.0 and 5.0, (2) application of 3 mM MgATP to the cytoplasmic side at pHi 7.0 or at the acidic pHi depending on the mutant form, and (3) application of 10 μg/ml of pLL for the complete inhibition of TREK-2, confirming the baseline of recording.
The WT hTREK-2 showed prominent activation by pHi 6.0 and further activation by pHi 5.0. The application of MgATP induced reversible inhibition (Fig. 2A). In contrast, a large basal inward K+ current (tonic activity) was observed in G334A. Responses to acidic pHi was the opposite of WT (Fig. 2B). Although not shown here, the application of MgATP at the neutral pHi 7.0 induced no significant inhibition in G334A as demonstrated in our previous study [15].
Then, we investigated the properties of the newly induced mutants combined with G334A. The K330A/G334A showed a tonic activity that was not activated further but slightly inhibited by acidic pHi. Also, the application of MgATP had no significant effect at pHi 7.0. (Fig. 2C). The G334A/R355-7A showed low basal activity and prominent activation by acidic pHi. At pHi 7.0, interestingly, the application of MgATP induced an initial inhibition followed by a slow activation that was not reversed but further enhanced by washout (Fig. 2D). The summary of the responses of the various hTREK-2 mutants to acidic pHi is shown as the bar graphs (Fig. 2E). The difference in response to MgATP between mutants except G334A/R355-7A was shown as a bar graph. The G334A/R355-7A is discussed in Fig. 3 (Fig. 2F).
Finally, we investigated whether the additional mutations of the distal three consecutive Arg residues abolished the persistent activation by acidic pHi in G334A/R355-7A. Different from the hypothesis, the property of G334A/R355-7A/R377-9A was not distinguishable from G334A/R355-7A; low basal activity, prominent activation by acidic pHi, initial inhibition, and subsequent slow activation by MgATP (Fig. 3A). To further analyze the biphasic responses, MgATP was also applied at pHi 6.0 (data not shown), which revealed more prominent changes of the three phases; (1) initial inhibition, (2) slow activation, and (3) rebound-like activation by washout (Fig. 3B). The biphasic responses to MgATP were similarly observed when R355-7A were solely mutated (Fig. 3C). The summary of the responses to MgATP is shown as the bar graphs (Fig. 3D). Interestingly, there was a tendency of lower slow activation phase in R355-7A than G334A/R355-7A and G334A/R355-7A/R377-9A.
DISCUSSION
The relation between molecular structure and its function is an intriguing theme in a variety of proteins including ion channels. Among the amino acids comprising proteins, glycine (Gly) is the simplest amino acid having a single hydrogen atom as its side chain. The lack of a side chain makes Gly the most flexible amino acid. Due to its small and compact form, Gly is often integral to the packing formation of alpha-helices in the transmembrane domain proteins. Also, it facilitates helix kinking that increases the contact between helices [16]. Also, together with proline (Pro), Gly could provide structural flexibility of the protein [17].
Owing to the smallest size, changing Gly to other amino acids with a larger side chain could break the structure of proteins. For this reason, we tried to analyze the dual mutants. Our present study shows that the combined mutation of G334 with K330 or with R355-7 induces changes in the basal activity and the responses to the acidic pHi and ATP, which were the same as the changed properties of K330A and R355-7A. Therefore, it is not likely that the substitution of Gly with Ala (G334A) extensively change the structure but only partly changed the balance between K330 and R355-7 with regard to the PIP2-dependent regulation. Although the experimental findings are indirect and circumstantial, we cautiously interpret that the G334 residue could actually play a hinge- or fulcrum-like role between the inhibitory (K330) and stimulatory (R355-7) cationic residues in the Ct (Fig. 1).
The second question of this study was a suspicious hidden role of the more distal triple Arg (R377-9) in the Ct of hTREK-2. Since both the R355-7A and the dual mutation (G334A/R355-7A) contained a similar capacity of activation by acidic pHi, we supposed that the more distal R377-9 might provide another binding site with anionic phospholipids. However, our present study indicates that R377-9 may not play a significant role even after neutralizing the R377-9. Therefore, the hypothesis of R377-9 as the second binding site was rejected. Nevertheless, since the relatively distal triple cationic residues are also conserved in the Ct of hTREK-1 (Fig. 1H), there might be still unknown regulatory function based on the electrostatic interaction, which requires future investigation.
An unexpected finding of the present study was the slow activation after the fast inhibition by ATP, a dual regulatory effect. The initial inhibition was consistent with our model of inhibitory interaction between PIP2 and K330, which has been consistently observed in the previous studies. However, there was no slow activation phase in the WT TREK-2 (Fig. 2A). But, slow activation phase was observed in R355-7A, G334A/R355-7A, and G334A/R355-7A/R377-9A (Figs. 2, 3). Although statistically insignificant, the extent of slow activation appeared more prominent in the G334A/R355-7A and G334A/R355-7A/R377-9A than R355-7A (Fig. 3D). In the experiment of the ATP effect, another notable finding was the rebound-like activation in response to the washout of ATP (Fig. 3). Together with the initial inhibition by ATP, it could be interpreted that the rebound-like activation might reflect the fast recovery from the inhibitory effect while the slower activation was not reversed yet. Taken together, the results suggested two independent mechanisms for the fast inhibition and slow activation by ATP in the proximal Arg (R355-7) mutants. Although the PIP2-dependent mechanism of the inhibitory effect is relatively well understood, the precise mechanism of slow activation is totally unknown yet. Nevertheless, since the slow activation was observed only in the site-directed artificial mutation of hTREK-2, the motivation for further investigation is not high.
The molecular structure of TREK-1 channel including the Ct was recently reported by Lolicato et al. [18]. The proximal region of Ct shows a long alpha-helix closed located to the inner leaflet of the plasma membrane. The putative bent conformation at Gly residue corresponding to G334 in hTREK-2 was not emphasized, and the possibility of Gly as the molecular fulcrum or hinge for the opposite regulation between K330 and R355-7 was not discussed in the study by Lolicato et al. [18]. According to our recent study and the present findings, the tonic high activity of G334A may be due to the putatively rigid conformation in the mutated alpha-helix of Ct. Based on the interpretation, we assumed that the flexibility or bent conformation at G334 may allow the K330-dependent inhibitory regulation with ATP in hTREK-2 (Fig. 1). Consistent with the hypothetical model, the combined mutation of K330A/G334A showed a tonic active state as observed in the K330A of hTREK-2 (Fig. 2).
In summary, the present study of hTREK-2 and its various mutants could provide further, although indirect, evidence supporting our model of the dual regulation by PIP2 via closely located cationic residues. The tug-of-war or see-saw model of Ct might be helpful to understand the complex responses of TREK channels since the allosteric interaction between proximal Ct and the fourth transmembrane segment is critical for the gating of TREK channels [19].
 XML Download
XML Download