

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Aripiprazole is a quinolinone derivative with partial agonist activities at dopamine D2 and 5-HT1A receptors [1,2]. It was approved as an atypical antipsychotic drug for the treatment of schizophrenia and bipolar mood disorder. It also exhibits antagonist activity at 5-HT2A receptor [3]. The efficacy of the major metabolite, dehydroaripiprazole, is comparable to that of the parent drug [4]. Partial agonist activity at D2 receptors has led to the concept of aripiprazole as a “dopamine system stabilizer”, decreasing the abnormally high level of dopamine activity, and increasing dopamine activities when they are abnormally low [1,5,6]. Aripiprazole has been considered as a drug of choice in populations at risk of QTc prolongation based on clinical studies revealing a low incidence of metabolic side effects, extrapyramidal symptoms and serious cardiac adverse effects [1]. Although most antipsychotics exhibit an unfavorable cardiovascular adverse effect profile, aripiprazole is associated with a low risk of sinus tachycardia, orthostatic hypotension, and QTc prolongation [7]. Despite the increasing use of aripiprazole, however, the pharmacological mechanism of aripiprazole is still under investigation and drug safety has yet to be established in pediatric populations as well as in the elderly with dementia, especially following long-term repetitive use. For example, aripiprazole can cause severe arrhythmia in a child [8]. QTc prolongation with aripiprazole has been reported in some patients [9,10].
In patch-clamp studies, aripiprazole inhibits human ether-a-go-go-related gene (hERG) K+ channels, which are fast delayed rectifier K+ currents with a potency (pEC50) of approximately 6.22 (~0.6 μM) [11]. However, this electrophysiological finding has yet to be replicated in multiple studies and in other ion channels such as voltage-gated K+ (Kv) channels critical for the repolarization of cardiac and neuronal action potentials (APs). A-type Kv channels mediate rapidly activating and inactivating transient outward currents in various tissues, including cardiac myocytes and neurons [12,13]. The Kv1.4 channels are activated at a low voltage followed by fast inactivation and slow recovery from inactivation. This electrophysiological property of Kv1.4 is associated with the regulation of AP duration and firing frequency in central nervous system, peripheral nervous system, and cardiac tissue. The Kv1.4 activity entails various physiological roles including motivation for reward, DRG nociception, and arrhythmia [14-16].
We determined the electrophysiological basis for the cardiac and neuronal actions of aripiprazole, by investigating the effects of aripiprazole on Kv1.4 currents expressed in HEK293 cells using a whole-cell patch-clamp technique. Aripiprazole exerts a concentration-dependent inhibition on Kv1.4 currents by preferentially interacting with the open state of the channel and also inhibits Kv4.3 currents, which are the other A-type Kv channels. The results of the present study may provide an ionic basis for the various cardiac and neurological effects of aripiprazole.
Go to : 
METHODS
Stable cell line generation and cell culture
A hKv1.4 stable cell line was generated in HEK293 cells by cloning human Kv1.4 cDNA (GenBank number: NM_002233) into pCDH-CMV-MCS-EF1α-Puro vector (System Biosciences, Palo Alto, CA, USA) to obtain hKv1.4-expressing plasmid pCDH-hKv1.4. HEK293 cells were transfected with pCDH-hKv1.4 vector using Lipofectamine 2000 (Life Technologies, Carlsbad, CA, USA) according to the manufacturer's recommendations. Positive clones were selected by the addition of puromycin (Life Technologies, Grand Island, NY, USA) to media. The hKv1.4 stable cell line was established by single colony isolation and characterization of the cellular electrophysiology. The hKv1.4-HEK293 stable cell line was grown in Dulbecco's modified Eagle's medium (Life Technologies, Grand Island, NY, USA) containing 10% fetal bovine serum at 37°C in a 5% CO2 atmosphere. The cells were plated on sterile glass coverslips placed inside a 35 mm culture dish. One day later, hKv1.4 currents were recorded using patch-clamp technique. A stable CHO cell line expressing Kv4.3 channels was maintained in Iscove’s modified Dulbecco’s medium (Gibco, Life Technologies Co., Grand Island, NY, USA) supplemented with 10% fetal bovine serum (Gibco), 1% HT supplement (Gibco), and 0.3% geneticin (Gibco), as described previously [17]. The cells were treated with trypsin-EDTA solution (Gibco) and seeded on glass coverslips (12 mm diameter) in 35 mm Petri dishes 1 day prior to use in patch-clamp recording.
Electrophysiology
Kv1.4 and Kv4.3 currents were recorded using the whole-cell patch-clamp technique with a Multiclamp 700B amplifier (Molecular Devices, San Jose, CA, USA) as described previously [18]. Data acquisition was performed with pClamp 10.0 software (Axon Instruments, Inc., Forster City, CA, USA) using a digidata1322A interface (Molecular Devices). The recording chamber (RC-26G; Warner Instruments, Hamden, CT, USA) was continuously perfused with an extracellular bath solution using gravity-fed flow (1 ml/min) at room temperature (22°C–24°C). Patch pipettes (3–4 MΩ) were pulled from borosilicate glass (1B150F-4; World Precision Instruments, Sarasota, Fl, USA) and filled with an internal solution containing (in mM) 140 KCl, 1 CaCl2, 1 MgCl2, 10 EGTA, and 10 N‐2-hydroxyethylpiperazine-N′‐2‐ethanesulfonic acid (HEPES, pH 7.3 with KOH). The external bath solution contained (in mM) 140 NaCl, 5 KCl, 1 CaCl2, 1 MgCl2, 10 glucose, and 10 HEPES (pH 7.3 with NaOH). The current signal was sampled at 20 kHz and filtered at 5 kHz. The series resistances were usually compensated up to 80% by amplifier circuits and no leak subtractions were used. Kv1.4 and Kv4.3 currents were evoked by applying 500 ms depolarizing pulses from a holding potential of –80 to +40 mV every 20 sec and 10 sec, respectively.
Aripiprazole and dehydroaripiprazole (MedChem Express, Monmouth Junction, NJ, USA) were dissolved in dimethyl sulfoxide (DMSO) as the stock solution. The stock solution was then further diluted with an external bath solution. The concentration of DMSO in the final dilution was less than 0.1% except for the concentration of 30 μM aripiprazole or dehydroaripiprazole in bath solution (0.3% DMSO). These concentrations of DMSO had no effect on Kv currents (data not shown). All chemicals for bath and internal solution, and DMSO were obtained from Sigma-Aldrich Co. (St. Louis, MO, USA).
Data analysis and statistics
The Clampfit 10.0 (Molecular devices) and Origin 8.0 software (Origin Lab Corp., Northampton, MA, USA) were used for the data analysis. The concentration-dependent inhibition of Kv currents by aripiprazole was fitted to the Hill equation: f = 1 / [1 + (IC50 / [D])n], where f denotes the fractional inhibition at drug concentration [D], IC50 is the drug concentration for half-maximum inhibition, and n represents the Hill coefficient. The current activation curves were fitted with the Boltzmann equation : G/Gmax = 1 / [1 + exp (–(V–V1/2) / k)], where V, V1/2, k, G and Gmax represent the test potential, potential at half-maximal current, the slope factor, the conductance, and the maximal conductance, respectively. The conductance was calculated using the formula, G = I / (V–EK), where I denotes Kv current amplitude and EK is the calculated equilibrium potential (–85 mV). Voltage dependence of inhibition by aripiprazole was plotted as a function of the membrane potential. The fractional inhibition by aripiprazole ranging between +20 and +60 mV was fitted with the Woodhull equation [19] : f = [D] / {[D] + KD(0) × exp(–zδFV / RT)}, where KD(0) denotes the apparent affinity at 0 mV (the reference voltage), z is the charge valence of the drug (+1 for aripiprazole and dehydroaripiprazole), δ represents the fractional electrical distance (i.e., the fraction of the transmembrane electric field sensed by a single charge at the receptor site), F is the Faraday’s constant, R is the gas constant, and T is the absolute temperature. In the present study, 25.4 mV was used as the RT/F value at 22°C.
Data are expressed as mean ± standard error. Statistical comparisons were performed with Student’s t-test and analysis of variance followed by Bonferroni’s test for comparisons of multiple groups using Excel and Origin 8.0 software. A value of p < 0.05 was considered statistically significant.
Go to :
RESULTS
Concentration-dependent inhibition of Kv1.4 by aripiprazole
Kv1.4 current was recorded with the whole-cell patch-clamp technique and its inhibition by aripiprazole was tested. Kv1.4 currents were evoked by 500-ms depolarizing pulses to +40 mV from a holding potential of –80 mV every 20 sec with or without aripiprazole (Fig. 1). Kv1.4 currents were activated and inactivated rapidly in response to membrane depolarization pulses, as described previously [20]. Extracellular application of aripiprazole (0.3 to 30 μM) resulted in moderate reduction of the peak amplitude and acceleration of decay phase of Kv1.4 currents in a concentration-dependent manner (Fig. 1A). The integral of the current during depolarization was measured to quantify the inhibition of Kv1.4 currents. The inhibition measured on the integral of Kv1.4 was fitted by Hill equation and yielded an IC50 value of 4.4 ± 0.4 μM with a Hill coefficient of 2.5 ± 0.2 (n = 8) (Fig. 1B). The concentration-response curve for Kv1.4 was rather steep with a Hill coefficient of 2.5, which suggested more than one binding sites for aripiprazole or a positively cooperative binding mechanism [21]. The time course for the decay of Kv1.4 at +40 mV under control conditions was fitted with a biexponential function with fast and slow time constants of 39.6 ± 3.8 ms and 184.9 ± 25.0 ms, respectively (n = 8) (Fig. 1C). With the application of aripiprazole, the time course for the decay of Kv1.4 was also fitted with a biexponential function. Aripiprazole decreased both the fast and slow time constants (12.3 ± 1.5 ms and 85.3 ± 2.8 ms, respectively, at 10 μM aripiprazole) in a concentration-dependent manner, suggesting that the inactivation kinetics of Kv1.4 were accelerated by aripiprazole. To investigate the effect on the activation kinetics, we measured the time-to-peak values of Kv1.4 currents (Fig. 1D). Under control conditions, the time-to-peak was 5.2 ± 0.2 ms (n = 8). In the presence of aripiprazole, the time-to-peak of Kv1.4 activation (3.7 ± 0.1 ms at 10 μM aripiprazole) was significantly reduced in a concentration-dependent manner. These accelerations in both decay rate and activation time courses of Kv1.4 suggests open-channel block mechanisms [22,23].
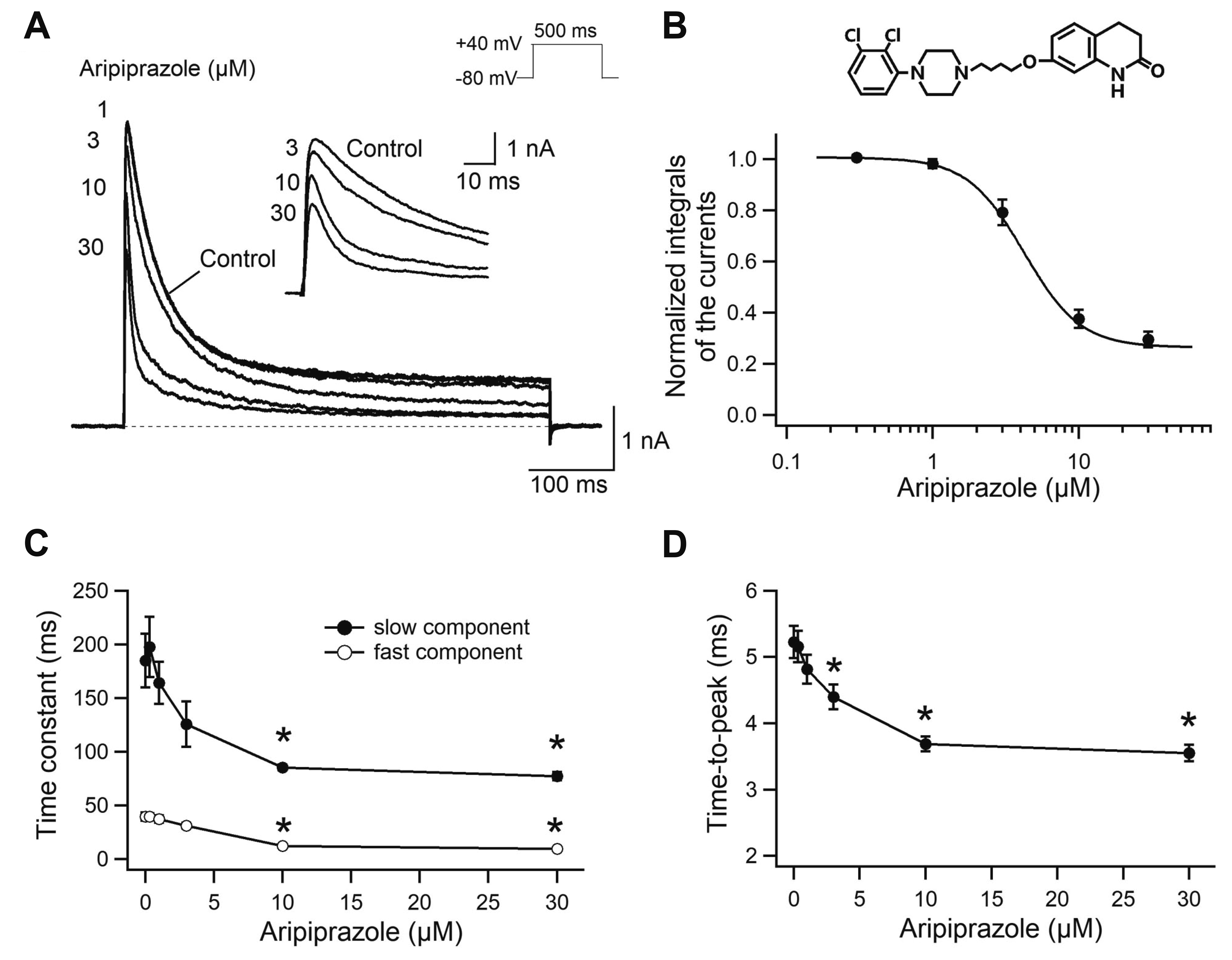 | Fig. 1Concentration-dependent inhibition of the Kv1.4 current by aripiprazole.(A) Representative Kv1.4 currents following the application of aripiprazole. Kv1.4 currents were evoked by 500-ms depolarizing pulses to +40 mV from a holding potential of –80 mV every 20 sec. The effects of 1, 3, 10, and 30 μM of aripiprazole are shown. The dotted line represents zero current. The inset shows first 60 ms of the traces with expanded time scale. (B) Normalized integrals of Kv1.4 currents during depolarization are plotted as a function of aripiprazole concentrations (n = 8). Data were fitted with the Hill equation (solid line). The chemical structure of aripiprazole is shown in the upper panel. (C) Inactivation kinetics (n = 8). The fast and slow components of time constants were obtained by biexponential curve fitting. (D) Time-to-peak of Kv1.4 currents (n = 8). Time constants and time-to-peak values as a function of aripiprazole concentration are shown. Data are expressed as the mean ± standard error. *p < 0.05 compared with the control.
|
Voltage-dependent inhibition of Kv1.4
The effects of voltage on the aripiprazole-induced inhibition of Kv1.4 were investigated by applying a series of 500-ms depolarizing pulses between –70 and +60 mV from a holding potential of –80 mV with and without 10 μM aripiprazole (Fig. 2A). Aripiprazole inhibited the amplitude of Kv1.4 currents at all membrane potentials. Normalized conductance (G/Gmax) at every membrane potential tested was plotted (Fig. 2B). The data were fitted to Boltzmann equation. The activation curve of Kv1.4 yielded the half-activation potential (V1/2 = –14.0 ± 4.0 mV, n = 8) and the slope factor (k = 22.6 ± 1.4) under control conditions. In the presence of aripiprazole, both half-activation potential (V1/2 = –17.3 ± 7.0 mV, p = 0.436) and slope factor (k = 24.1 ± 1.1, p = 0.413) of activation curve were unchanged. To analyze the voltage-dependence of inhibition by aripiprazole, the fractional block of the integral (Iaripiprazole/Icontrol) was plotted as a function of membrane potential (Fig. 2C). The aripiprazole-induced inhibition was steeply increased in a voltage range corresponding to that of Kv1.4 channel activation (F4,35 = 3.37, p < 0.05). The inhibition was also voltage-dependent at more positive potentials (+20 to +60 mV) where the channels were fully activated. This voltage dependence of aripiprazole-induced inhibition was fitted to a Woodhull’s equation: δ = 0.17 ± 0.05 (n = 8).
 | Fig. 2Voltage-dependent inhibition of Kv1.4 by aripiprazole.(A) Kv1.4 currents were evoked by a series of 500-ms depolarizing pulses between –70 and +60 mV from a holding potential of –80 mV in the absence and in the presence of 10 μM aripiprazole. The inset shows first 60 ms of the traces with expanded time scale. (B) Plots of normalized conductance (G/Gmax) versus tested membrane potentials (n = 8). Conductance (G) was calculated by dividing the peak amplitude of Kv1.4 at the test potential by assuming a reversal potential of –85 mV under the ionic conditions of our experiment. The maximal conductance (Gmax) was obtained by fitting the normalized data using a Boltzmann equation. (C) Integral currents of Kv1.4 in the presence of aripiprazole were normalized to those of the control at each voltage (n = 8). Dashed line represents the activation curve of Kv1.4 under control condition. Voltage dependence was fitted to Woodhull’s equation (see Methods): δ = 0.17 ± 0.05 (n = 8). The solid line represents the linear fit. *p < 0.05 compared with the value obtained at –30 mV.
|
Effects on the recovery from inactivation and use dependence of Kv1.4
Recovery from inactivation of Kv1.4 was studied using a two-pulse protocol (Fig. 3). The cell was depolarized to +40 mV for 500 ms by prepulse and then repolarized to –80 mV holding potential for a variable period ranging from 0.3 sec to 14 sec before applying the same test pulses in the absence and presence of 10 μM aripiprazole (Fig. 3A). The peak amplitudes of the test pulse were normalized to those of prepulse and plotted against the inter-pulse intervals. In the control experiments, the time course of recovery from inactivation of Kv1.4 was fitted to a single exponential function with a time constant of 4.9 ± 0.8 s (n = 7) (Fig. 4B). After application of aripiprazole, the recovery from inactivation was also fitted to a single exponential function with a time constant of 5.5 ± 0.7 sec (n = 7, p = 0.105), suggesting that aripiprazole has no effect on the time course of recovery from inactivation of Kv1.4.
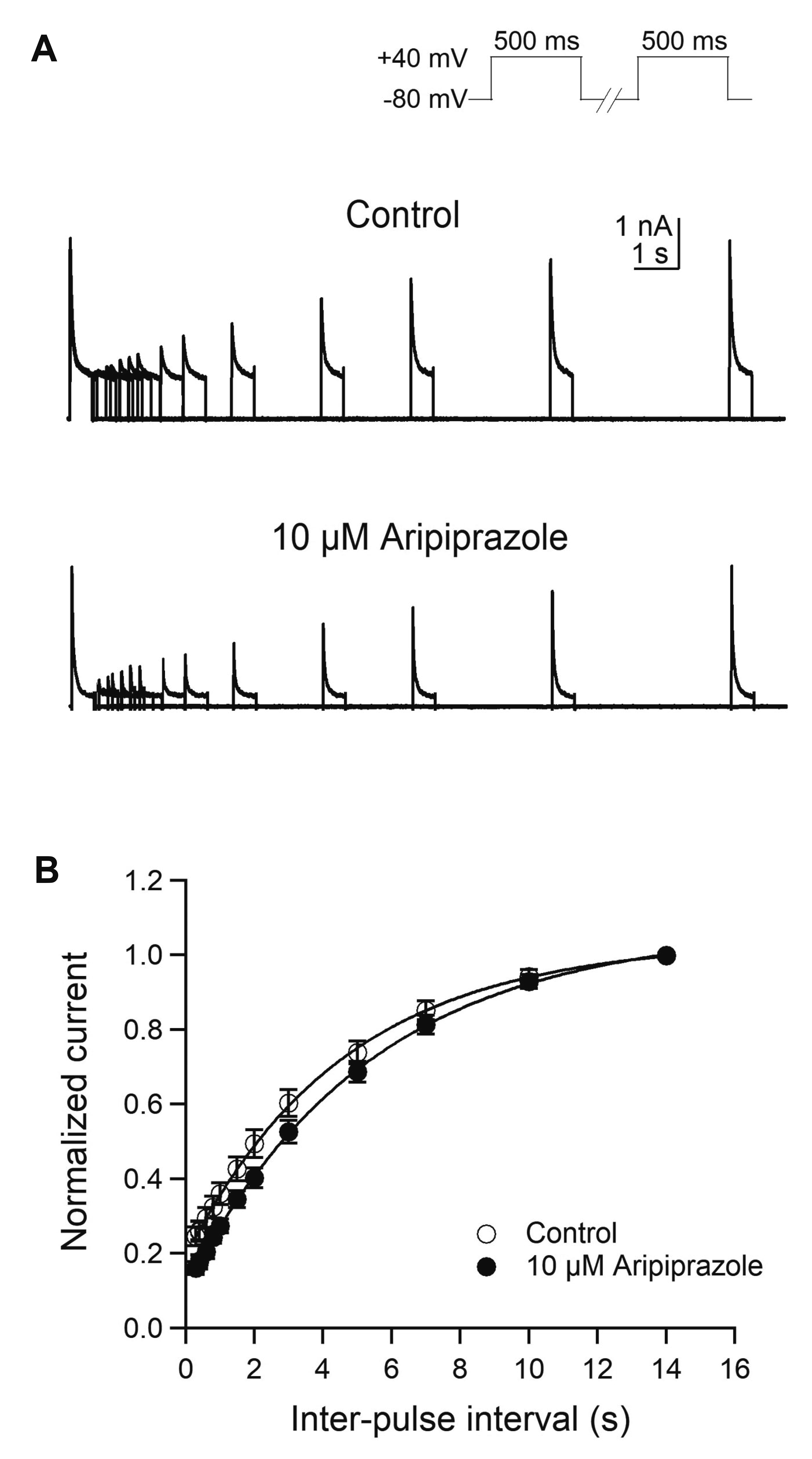 | Fig. 3The effect of aripiprazole on the recovery from inactivation of Kv1.4 current.(A) Representative current traces of Kv1.4 were recorded using two-pulse protocols with varying inter-pulse intervals in the absence and presence of 10 μM aripiprazole. (B) Time course of recovery from inactivation of Kv1.4 was fitted with a single exponential function (n = 7). The peak amplitudes of the test pulse were normalized to those of pre-pulse and plotted against the inter-pulse intervals.
|
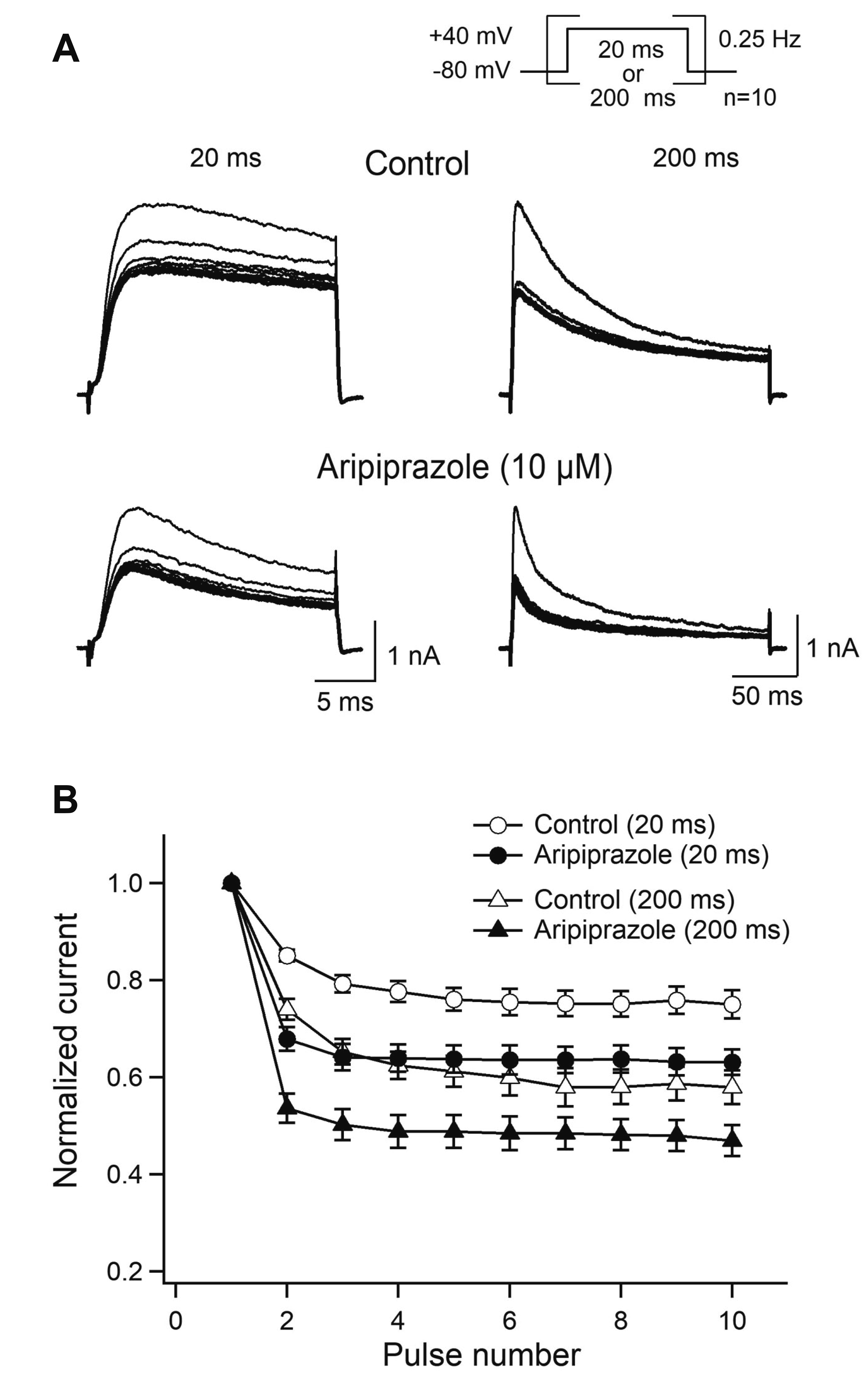 | Fig. 4Use-dependent inhibition of Kv1.4 by aripiprazole.(A) Ten repetitive 20- or 200-ms depolarizing pulses were applied to +40 mV from a holding potential of –80 mV at a frequency of 0.25 Hz under control conditions and after the application of 10 μM aripiprazole. (B) The peak amplitudes of the current at each pulse were normalized to those of the current obtained at the first pulse and then plotted as a function of the pulse number (n = 6).
|
To analyze the use-dependent inhibition by aripiprazole, Kv1.4 currents were evoked by 10 consecutive 20-ms and 200-ms pulses to +40 mV from a holding potential of –80 mV at 0.25 Hz in the absence and presence of aripiprazole (Fig. 4A). Since the peak amplitudes of the Kv1.4 currents were diminished to less than 50% by repetitive pulses at 0.5–2 Hz under control conditions (data not shown), we analyzed the use-dependent inhibition with aripiprazole at 0.25 Hz under two pulse durations (20 and 200 ms). Under control conditions, the Kv1.4 currents decreased by 25.0% ± 2.9% and 36.8% ± 2.6% at 20-ms and 200-ms pulses, respectively, after 10th repetitive pulses (n = 6) (Fig. 4B). In the presence of aripiprazole, the Kv1.4 currents decreased by 42.2% ± 3.5% (p < 0.001) and 53.1% ± 3.2% (p < 0.001) at 20-ms and 200-ms pulses, respectively. However, the calculated percentages of inhibition by aripiprazole were similar between 20 ms (17.2%) and 200 ms pulses (16.3%) (p = 0.267). These results suggest that aripiprazole exhibits use-dependent inhibition with successive pulses on Kv1.4 currents.
Concentration-dependent inhibition of Kv4.3 by aripiprazole
Next, we investigated the effect of aripiprazole on another voltage-gated A-type Kv channel, Kv4.3 (Fig. 5). Kv4.3 currents expressed in CHO cells were evoked by the same voltage protocol as in Kv1.4 currents except for the 10-sec interval in voltage step and were measured under control conditions and in the presence of various concentrations of aripiprazole (0.3 to 30 μM). Under control conditions, the Kv4.3 currents were activated to a peak and inactivated rapidly as reported previously [17], which was similar to those of Kv1.4 in Fig. 1A. In the presence of aripiprazole, the peak amplitude of Kv4.3 currents also decreased (Fig. 5B). As shown in Kv1.4 currents, to quantify the inhibition of Kv4.3 currents, the integral of the currents during the entire depolarization pulse was measured. The normalized inhibition of Kv4.3 currents plotted as function of drug concentration was fitted by Hill equation and yielded an IC50 value of 4.9 ± 0.4 μM (n = 5, p = 0.580 compared with Kv1.4) with a Hill coefficient of 2.3 ± 0.2 (p = 0.463 compared with Kv1.4) (Fig. 5B). These results suggest that aripiprazole showed similar inhibitory effects on both A-type Kv channels, Kv1.4 and Kv4.3.
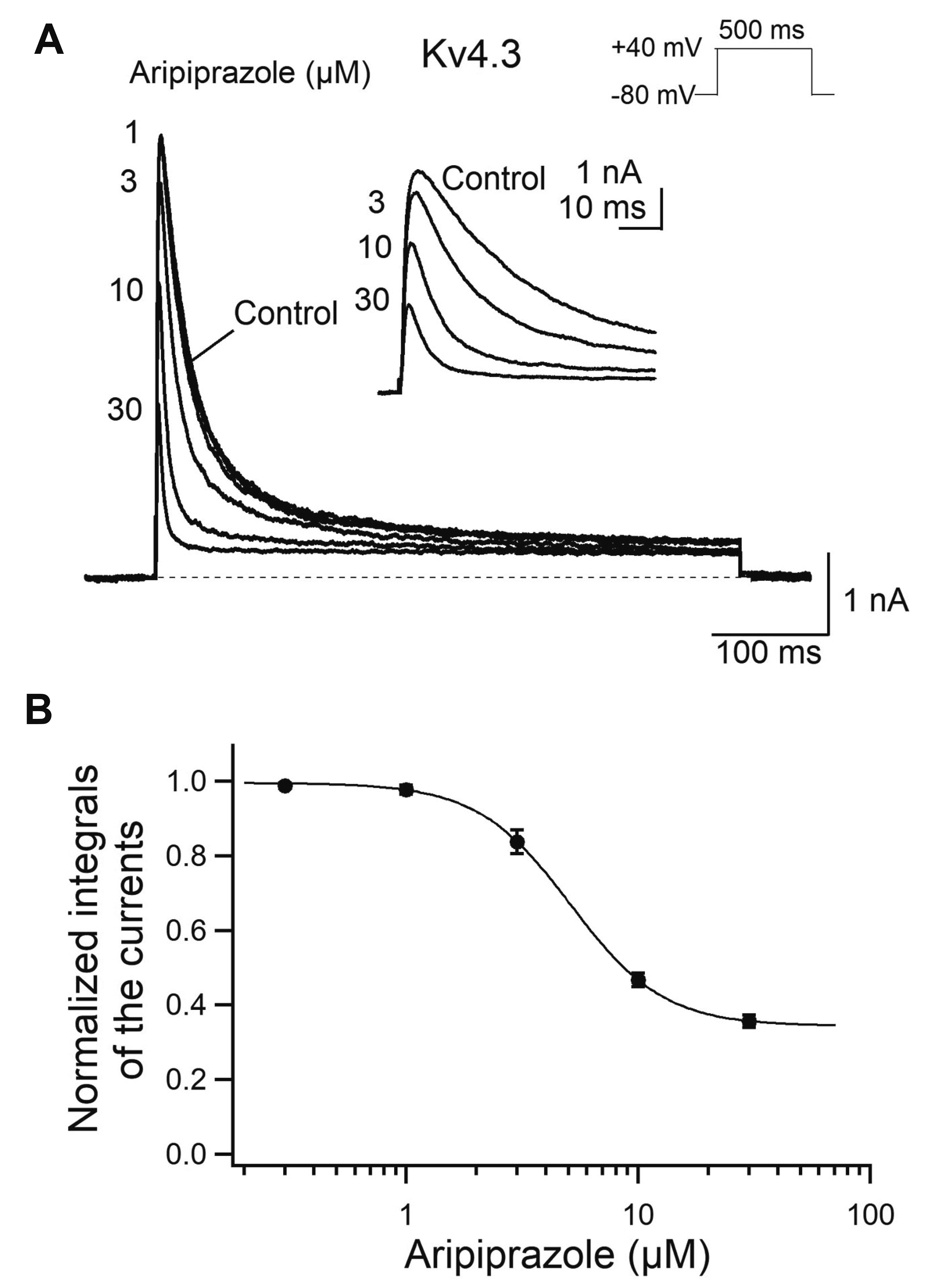 | Fig. 5Concentration-dependent inhibition of the Kv4.3 current by aripiprazole.(A) Kv4.3 currents were evoked by a 500-ms depolarizing pulse to +40 mV from a holding potential of –80 mV every 10 sec. The effects of 1, 3, 10, and 30 μM aripiprazole are shown. The dotted line represents zero current. The inset shows first 60 ms of the traces with expanded time scale. (B) Normalized integrals of Kv4.3 currents during depolarization are plotted as a function of aripiprazole concentrations (n = 5). Data were fitted with the Hill equation (solid line).
|
Effects of dehydroaripiprazole on Kv1.4 currents
A major active metabolite of aripiprazole, dehydroaripiprazole, has been reported to exhibit similar pharmacological activity compared to the parent compound [6,24]. To analyze the effect of dehydroaripiprazole on Kv1.4 currents, a series of dehydroaripiprazole concentrations were tested (Fig. 6). Bath application of dehydroaripiprazole reduced the integral of the Kv1.4 currents in a concentration-dependent manner (Fig. 6A). The normalized Kv1.4 currents plotted as a function of drug concentration was fitted to Hill equation (Fig. 6B) and yielded an IC50 value of 6.3 ± 0.8 μM (n = 5, p < 0.05 compared with aripiprazole) with a Hill coefficient of 2.0 ± 0.2 (p = 0.185 compared with aripiprazole). These results suggest that dehydroaripiprazole has inhibitory effects comparable to the parent compound, aripiprazole.
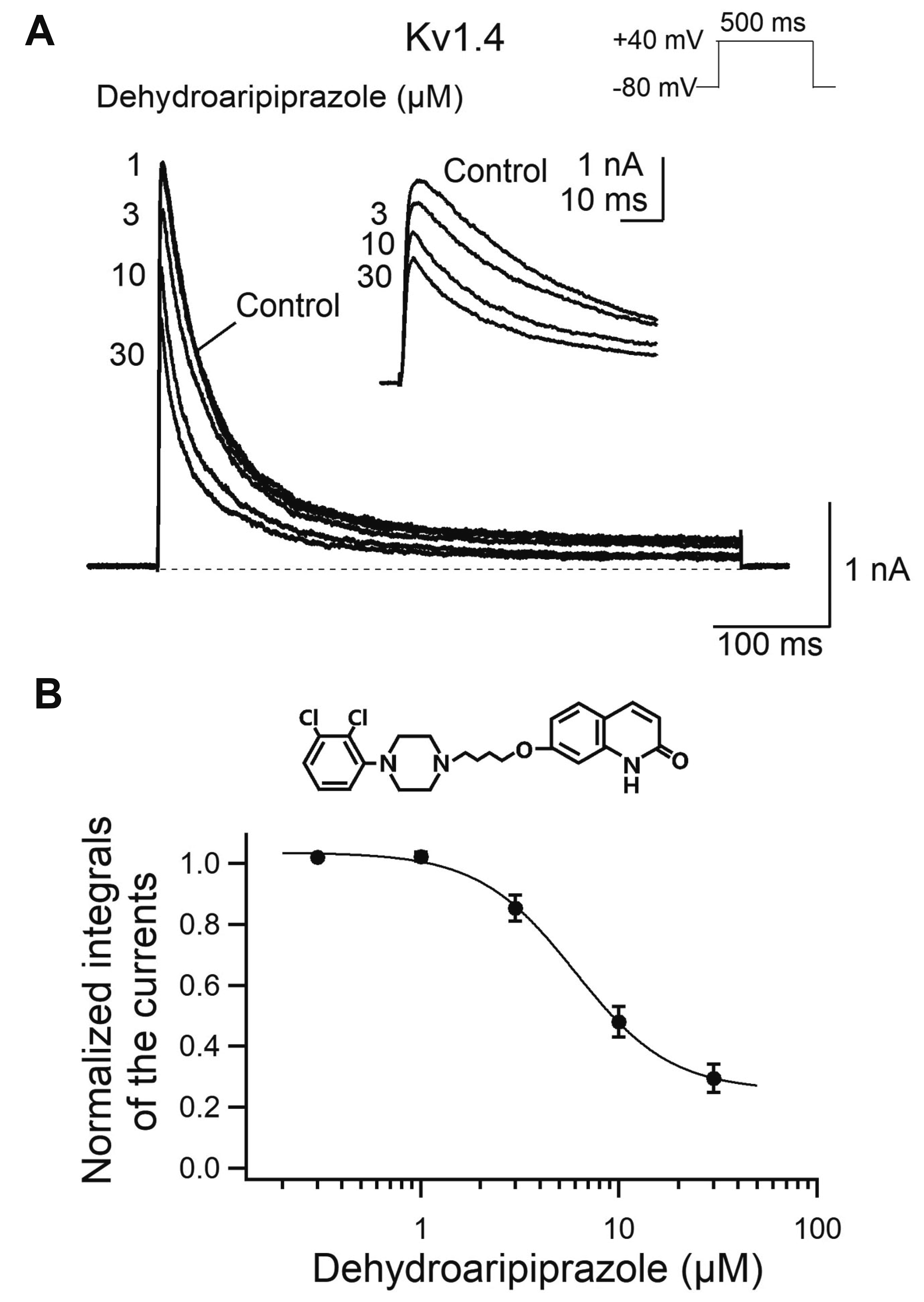 | Fig. 6The effect of dehydroaripiprazole, an aripiprazole metabolite, on the Kv1.4 current.(A) Kv1.4 currents were evoked by a 500-ms depolarizing pulse to +40 mV from a holding potential of –80 mV every 20 sec in the absence and presence of dehydroaripiprazole (1–30 μM). The dotted line represents zero current. The inset shows the Kv1.4 currents on an expanded time scale for the first 60 ms of the traces. (B) Normalized integrals of Kv1.4 currents during depolarization are plotted as a function of dehydroaripiprazole concentrations (n = 5). Data were fitted with the Hill equation (solid line). The chemical structure of aripiprazole is shown in the upper panel.
|
Go to :
DISCUSSION
We studied the effect of aripiprazole on Kv1.4 channels, A-type K+ channels, using whole-cell patch-clamp technique. The main findings are: 1) aripiprazole decreased peak amplitude of Kv1.4 currents and accelerated the inactivation kinetics in a concentration-dependent manner and 2) inhibition of Kv1.4 currents by aripiprazole was voltage- and use-dependent. Furthermore, the major metabolite dehydroaripiprazole showed similar inhibitory effects against Kv1.4. Aripiprazole similarly decreased another A-type K+ channels Kv4.3 in a concentration-dependent manner. Thus, the effects of aripiprazole on these currents may be explained by the inhibition of A-type K+ channels in the open states.
In the present study, aripiprazole inhibited A-type K+ currents in HEK cells stably expressing Kv1.4 channels, with decreasing activation time and accelerating inactivation kinetics. In contrast, aripiprazole failed to change the activation curves of Kv1.4 channels. It also induced a voltage-dependent block of Kv1.4 currents over the voltage range of activation (+20 to +60 mV). All these findings indicate the preferential binding of aripiprazole to the open state of the channel [25]. Aripiprazole shortened the time to peak and decreased the amplitude of Kv1.4 currents. The instantaneous inhibition following depolarization could be attributed to an interaction of drugs with the closed state of the channel. However, further inhibition during the depolarization suggests that aripiprazole also interacted with the open channel [22]. Furthermore, it is unlikely that the acceleration of current inactivation was due to altered gating kinetics because activation properties of Kv1.4 are not affected by aripiprazole (Fig. 2). Previously, we also demonstrated that fluoxetine blocked Kv1.4 currents with preferential binding to the open state in CHO cells expressing rat Kv1.4 channels [20].
In the present study, the rate of inactivation of Kv1.4 was a biexponential process, consistent with previous studies [26,27]. The fast and slow components may represent N-type and C-type inactivation, respectively [27]. Aripiprazole accelerated two types of inactivation rates. Fast activation and inactivation characterize A-type K+ channels. It is known that Kv1.4 channels, in contrast to all other Kvα subunits, consist of two inactivation domains at its N-terminus [28]. Inactivation domain 1 (ID1) at residues 1–38 and inactivation domain 2 (ID2) at residues 40–68 comprise small hydrophobic amino acids [29,30]. It is known that N-type inactivation and intracellularly acting open channel blockers are similar. Most open channel blockers such as aripiprazole are lipophilic with a positively charged nitrogen group. The receptor site for both drug and N-terminal binding has been localized to residues on the cytoplasmic side of S6 [31,32]. Thus, aripiprazole may inhibit Kv1.4 channels from the inside. In the present study, aripiprazole might be positively charged at an intracellular pH pf 7.3 because the drug is a very weak base with a pKa = 7.6 [33]. In the present study, aripiprazole exhibited voltage-dependent inhibition of Kv1.4 channels. The inhibition was apparent over the voltage range of activation (+20 to +60 mV). The voltage-dependent inhibition of Kv1.4 currents by aripiprazole can be explained by the interaction between the charged form of the drug and the binding sites in the electrical field of Kv1.4 channels. A fractional electrical distance (δ) calculated using the Woodhull model was 0.17 in this experiment with 10 μM aripiprazole. The δ value indicated that aripiprazole senses about 17% of the applied transmembrane electrical field as referenced from the intracellular side [34]. This finding reflects blockade of the open channel from the inside of the membrane [35,36].
Recovery from inactivation and use dependence of Kv channels are important properties contributing to the shape of repetitive APs. Use-dependent block is generally associated with a slow rate of recovery from inactivation due to the slower dissociation of the drug from its binding site [25]. Use-dependent block is accomplished by the binding of the drug either to the open or the inactivated states of channels [25]. In the present study, aripiprazole exhibited use-dependent inhibition of Kv1.4 channels, without affecting the recovery from inactivation. Furthermore, no change was found in the degree of block between the short- (20 ms) and the long-duration (200 ms) pulses in the presence of the drug. These findings exclude the possibility that the drug interacts with Kv1.4 channels in the inactivated state. Otherwise, the use-dependent inhibition may be explained by the competition between the modulatory site of inactivation and aripiprazole [25].
The serum aripiprazole concentration of the patients ranged from 173 to 367 ng/ml [4,37], which is equivalent to 0.4 to 0.8 μM. The maximal level of aripiprazole was 549 ng/ml (1.2 μM). The receptor binding profile of dehydroaripiprazole, which is a major active metabolite of aripiprazole, is similar to that of the parent compound, and therefore is expected to show similar pharmacologic activity [6,24,37]. Under steady state, the systemic exposure to the metabolite has been reported to represent 39% of that of the parent drug [4,38]. The plasma concentration of this metabolite may therefore be clinically relevant to both outcome and adverse effects, although the comprehensive effects have yet to be reported [39]. In the present study, dehydroaripiprazole exhibited similar inhibitory effects on Kv1.4 channels. Thus, long-term use of aripiprazole lead to the accumulation of dehydroaripiprazole, which inhibits Kv1.4 channels, similar to aripiprazole.
A-type K+ channels are critical to shape APs in neuronal cells and cardiac cells. The different types of A-type K+ channels are localized in neuronal structures with distinct distribution density depending on the cell types. Kv1.4 channels are located in the soma, axons and axon terminals while Kv4.3 are preferentially located in somatodendritic compartments in the central nervous system [40,41]. Kv1.4 is the only Kv1α subunit expressed in smaller diameter neurons of dorsal root ganglions, suggesting that homomeric Kv1.4 channels predominate in pain-related Aδ and C fibers arising from these cells [15]. Kv1.4 channels are also expressed in cortical pyramidal cells [42]. Kv4.3 is highly expressed in distal apical dendrites of cortical pyramidal cells [43], which are critical in determining the dendritic spread of backpropagating APs [44]. Thus, aripiprazole may inhibit Kv1.4 and Kv4.3 channels, which may alter synaptic transmission and the shape and spread of APs in both peripheral and central nervous system. In the heart, there are two types of transient outward currents based on their voltage-dependent kinetics of recovery from inactivation [45]. The two phenotypes including Ito,fast and Ito,slow are mediated by Kv4.2/4.3 and Kv1.4 channels, respectively. It is interesting that aripiprazole similarly inhibits Kv1.4 and Kv4.3 channels characterized by different inactivation mechanisms, rates of recovery from inactivation and subcellular localizations in various neuronal cells. In addition to the inhibition, the use-dependent blockade of A-type K+ channels might be more important in burst firing of APs in neurons and in tachycardia.
Go to :
 XML Download
XML Download