

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The KCNQ family constitutes slowly-activating potassium channels among voltage-gated potassium channel superfamily [1]. One of the most well-recognized physiological roles vested by KCNQ family would be conduction of slowly-activating delayed rectifier K current (IKs) in ventricular myocyte carried by KCNQ1 (KvLQT1, Kv7.1) and its ancillary subunit, minK (KCNE1). Loss-of-function mutation in KCNQ1 may lead to detrimental prolongation of phase II of cardiac action potential, hence longer QT interval in electrocardiogram (ECG) [2]. Even sudden cardiac arrest can be manifested if certain triggers such as hypercalcemia exist. In addition to KCNQ1, there are four other subunits in KCNQ family (KCNQ2–KCNQ5). KCNQ2 and KCNQ3 are well-known for their contribution to tonic suppressive effect to membrane excitability in neurons. Such membrane stabilization by KCNQ2 and 3 can be abruptly seized as soon as acetylcholine-muscarinic stimulation is given to the cell [3-5]. Loss-of-function mutation in KCNQ2 and KCNQ3 gene may lead to pathologic membrane excitability in neurons and such mutations are highly associated with benign familial neonatal epilepsy. Tissue-specific expression profiles and physiological roles of KCNQ family are well-documented and can be found in other extensive reviews [1,6-8].
Meanwhile, there have been consistent arguments about functionality of KCNQ4 and KCNQ5 channels in smooth muscle cells. Some have advocated that the membrane stabilizing effects by KCNQ4 and KCNQ5 channels in smooth muscle cells are substantial that both channels could be novel therapeutic targets for smooth muscle related disorders such as achalasia, asthma, chronic obstructive pulmonary disease (COPD), cerebral vasospasm, pulmonary arterial hypertension, preeclampsia, overactive bladder and others [6,9]. As disease entities suggest, KCNQ4 and KCNQ5 channels are implicated in various smooth muscle physiology such as gastrointestinal (GI) tract, vasculature, uterine/myometrium, sphincter muscle, and many others.
Notably, concordant relaxation of corpus cavernosum smooth muscle and surrounding arterial vascular smooth muscle have been well recognized as a core mechanism of penile erection [10-13]. It is also generally acknowledged that benign prostate hypertrophy and erectile dysfunction are highly correlated, hence such condition is called lower urinary tract syndrome (LUTS). Inhibition of phosphodiesterase 5 (PDE5) by sildenafil has been a promising therapeutic for erectile dysfunction and LUTS, however, many have reported that some patients show insufficient response to conventional drugs [11,13,14]. Recently, strong expression of KCNQ4 and KCNQ5 in corpus cavernosum was reported [14] and KCNQ-mediated corporal smooth muscle relaxation was suggested as a novel therapeutic pathway for erectile dysfunction, especially for sildenafil-refractory group.
As mentioned earlier, cardiac IKs is mediated by not only through KCNQ1 but also with minK (KCNE1), a β ancillary subunit. Pathognomonic mutations in minK alone are categorized as independent subtype of long QT syndrome (LQT5) suggesting that interaction between pore-forming α subunit and β ancillary subunit is not negligible [2,15]. In a sense, there have been a number of studies showing evidences for co-expression of KCNQ and KCNE in various tissues [16-19]. Some have shown functional implication of such KCNQ-KCNE interactions using electrophysiology in heterologous expression systems [20-23]. In male urinary tract, especially in corpus cavernosum, it was found that both transcription and protein expression level of KCNE4 were dominant among 5 KCNE subunits (KCNE1–KCNE5) [14].
In this study, we first synthesized a potent KCNQ4, 5-specific activator based on a reaction scheme suggested by Seefeld et al. [24]. We named this activator as URO-K10 and evaluated electrophysiological characteristics of URO-K10 activated KCNQ4 and KCNQ5 channels in HEK293 cells. We also created a series of fusion proteins so that KCNQ4, KCNQ5, and KCNE4 have additional fluorescence/epitope tags at their C-termini. Using such constructs, we evaluated protein-protein interaction between the subunits using Foster Resonance Energy Transfer (FRET) and co-immunoprecipitation (co-IP). Moreover, we evaluated electrophysiological consequences of KCNQ-KCNE interaction from URO-K10 activated whole-cell currents. Last but not least, we tested if URO-K10 can induce relaxation of mesenteric vascular smooth muscle, a tissue known for strong, endogenous co-expression of KCNQ4 and KCNE4 [17,25,26].
METHODS
Cell culture and transient transfection
Human embryonic kidney (HEK293) cells (ATCC, Manassas, VA, USA) were maintained according to the supplier’s recommendations. For the transient transfection for electrophysiology, the cells were seeded in 12-well plates. The following day, 0.5–1 g/well of cDNA was transfected using the transfection reagent FuGENE 6 (Roche Molecular Biochemicals, Indianapolis, IN, USA), as detailed in the manufacturer’s protocol. After 30–40 h, the cells were trypsinized and transferred to a small recording chamber for whole-cell recording. For the transient transfection for co-immunoprecipitation, the cells were seeded in 6-well plates. After two days, 1 g/well of cDNA was transfected using the transfection reagent Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA), as detailed in the manufacturer’s protocol. After 24 h, the cells were washed and lysed for further protocols.
Molecular cloning
All mutagenesis was conducted using QuikChange II XL site-directed mutagenesis kit (Agilent Technologies, La Jolla, CA, USA). Human KCNQ4 cDNA in pRK5-Myc vector was kindly donated by Dr. Heon Yung Gee (Yonsei University). Human KCNQ5 cDNA and human KCNE4 cDNA in pcDNA3.1(+)/C-(K)DYK vector were purchased from GenScript (Piscataway, NJ, USA). For subcloning and vector exchange, the coding sequence (CDS) of KCNQ4 construct was truncated with SalI and BamHI restriction enzymes. Likewise, the CDS of KCNQ5 construct was truncated with EcoRI and BamHI, and the CDS of KCNE4 construct was truncated with BglII and BamHI restriction enzymes. All truncated insert CDS was ligated into accordingly excised pECFP-N1, pEGFP-N1 or pEYFP-N1 vectors. pECFP-N1, pEGFP-N1 or pEYFP-N1 vectors were all purchased from Clontech (Takara Bio, Mountain View, CA, USA).
Electrophysiology
The cells were transferred to a small chamber on the stage of an inverted microscope (Eclipse Ti, Nikon, Tokyo, Japan) and attached to coverslips in the small chamber for 10 min prior to the patch recording. Whole-cell and single-channel currents were recorded room temperature using borosilicate patch pipettes of 1–3 MΩ and 15–35 MΩ respectively. High pipette resistance for single-channel current recording was achieved by further heat-microforging of the glass pipettes. The currents were recorded using an Axopatch 200B patch-clamp amplifier (Molecular Devices, Foster City, CA, USA). pClamp software v.10.2 or v.11.1 and Digidata 1440A (Molecular Devices) were used for data acquisition and application of the command pulses. Low-pass Bessel filter with 2 kHz cut-off frequency and 5 kHz sampling frequency were selected for non-kinetic analysis recordings with long recording duration. 5 kHz low-pass Bessel filter cut-off frequency and 10 kHz sampling frequency were selected for kinetic analysis recordings. For single-channel recording, 1 kHz low-pass Bessel filter cut-off frequency and 10 kHz sampling frequency were used. Pipette capacitance cancellation up to 90% was achieved in cell-attached modes of every recording. After whole-cell configuration, almost total cancellation of whole-cell capacitance was achieved especially in cells for kinetic analysis. Consequent manipulation of series resistance was necessary but compensation of the series resistance was not achieved on purpose for pipette resistance remained significantly low (~2 MΩ). The data were analyzed using pClamp v.10.2 or v.11.1 and Origin Pro 8 software (OriginLab, Northampton, MA, USA). For illustrations, single-channel data were digitally filtered (210 Hz lowpass Gaussian filter) and sampled at 2 kHz (data reduction factor = 5). Data with original time resolution were used for open probability analysis and unitary current calculation.
For bath solutions for whole-cell recordings of the KCNQ channels, we used Normal Tyrode solution (NT) unless otherwise mentioned. The Normal Tyrode solution contained 135 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 10 mM glucose, and 10 mM HEPES with a pH of 7.4 adjusted with NaOH. URO-K10 compound was synthesized by Sundia MediTech Company (Sanghai, China) based on reaction scheme suggested by Seefeld et al., [24] (Fig. 1). ML-213 was purchased from Tocris Bioscience (Bristol, UK) and XE-991 were purchased from Sigma Aldrich (St. Louis, MO, USA). All drugs were diluted with DMSO up to 1000x stock solution and each stock solution was further diluted using bath solutions. The internal solution contained 140 mM KCl, 10 mM HEPES, 0.2 mM Tris-GTP (Tris-guanosine 5’-triphospate), 4 mM Na-ATP (adenosine 5’-triphosphate), 10 mM EGTA, 5.37 mM CaCl2 ([Ca2+]free = 100 nM), and 1.75 mM of MgCl2 ([Mg2+]free = 1 mM) with a pH of 7.3 adjusted with KOH. For cell-attached single-channel recordings, filtered Normal Tyrode solution supplemented with either 1000x URO-K10 or DMSO were used as pipette solutions. Normal Tyrode solutions were continuously perfused in bath at all times.

Fig. 1
Chemical nature, reference, purchase information of the agonist/antagonists used in the study.
(1) URO-K10 was synthesized by Sundia MediTech Company (Sanghai, China) based on reaction scheme suggested by Seefeld et al. [24]. (2) ML-213 was purchased from Tocris Bioscience (Bristol, UK). (3) XE-991 was purchased from Sigma Aldrich (St. Louis, MO, USA).
![]()
Mathematical formulation
Voltage-dependency of steady-state conductances were analyzed by fitting the normalized conductance (G/Gmax)-membrane potential (V) data points into Boltzmann distribution function [1]. Namely,
where V1/2 is half-maximal voltage and k is a slope factor.
For the analysis of time-dependency of activation process at a given membrane potential, conductances were fitted to either single exponential function or double exponential function. Namely,
voltage-clamp at V0 yields,
hence,
Recall Hodgkin-Huxley model [27];
For KCNQ family shows remarkably slower activation process compared to delayed-rectifier K channels, we reduced the order of the exponential term to 1. The activation process of KCNQ4 channels were fitted well with first-order single exponential term, however, the activation process of KCNQ5 channels required two first-order exponential components. Namely,
where c1 and c2 are coefficients.
Dose-dependency of URO-K10 onto whole-cell current was analyzed by fitting each data points into dose-response function. Namely,
Open probability of the channel at given condition was analyzed as following. For a recording duration Tm at mth condition, total number of open events (no), closed events (nc), sum of each open dwell-time (τo) and each closed dwell-time (τc) were calculated respectively, from the idealized single-channel recording trace. Absolute current amplitudes for closed state and open state were manually assigned. Namely,
Microscopic image acquisition and FRET measurements
HEK293 cells were cultured in a 35-mm coverslip bottom dish or a 12-well plate to obtain images and measure FRET efficiency. To obtain the image and FRET efficiency of a cell, we used an inverted microscope with a 60x oil objective lens and the three-cube FRET calculation [28,29] controlled by MetaMorph 7.6 (Molecular Devices). The three-cube FRET efficiency (cube settings for CFP, YFP, and Raw FRET) was acquired from a pE-1 Main Unit to three-cube FRET (excitation, dichroic mirror, filter) through a fixed collimator: CFP (ET 435/20 nm, ET CFP/YFP/mCherry beam splitter, ET 470/24 nm, Chroma); YFP (ET 500/20 nm, ET CFP/YFP/mCherry beam splitter, ET 535/30 nm, Chroma); and CFP/YFP FRET (ET 435/20 nm, ET CFP/YFP/mCherry beam splitter, ET 535/30 nm, Chroma). The excitation LED and filter were sequentially rotated, the rotation period for each of the filter cubes was ~0.5 sec, and all images (three for CFP/YFP/Raw FRET) were obtained within 2 sec. Each of the images was acquired on a cooled 3 MHz (14 bit) EMCCD camera (iXon Ultra 888: ANDOR) with an exposure time of 100 ms with 1 × 1, 2 × 2, or 3 × 3 binning under the control of MetaMorph 7.6 software. Our FRET recording of the fluorophores was restricted in a range of CFP/YFP ratio from 0.5 to 2.0.
FRET Ratio (FR) and FRET efficiency computation
The FRET Ratio (FR) [29] is equal to the fractional increase in YFP emission due to FRET and was calculated as FR = FAD/FA = [SFRET(DA) − RD1 · SCFP(DA)]/(RA1 · [SYFP(DA)–RD2 · SCFP(DA)]). Here, SCUBE(SPECIMENDA) denotes an intensity measurement, where CUBE indicates the filter cube (CFP, YFP, or FRET), and SPECIMEN indicates whether the cell is expressing the donor (D; CFP), acceptor (A; YFP), or both (DA). RD1 = SFRET(D)/SCFP(D), RD2 = SYFP(D)/SCFP(D), and RA1 = SFRET(A)/SYFP(A) are predetermined constants from measurements applied to single cells expressing only CFP- or YFP-tagged molecules. Although three-cube FRET does not require that CFP and YFP fusion constructs preserve the spectral features of the unattached fluorophores, similar ratios and recorded spectra furnished two indications that the spectral features of the fluorophores were largely unperturbed by fusion. Since the FR relies on YFP emission, YFP should be attached to the presumed limiting moiety in a given interaction. Subsequent quantitative calculations based on FR relied on a presumed 1:1 interaction stoichiometry. The effective FRET efficiency (EEFF) was determined by EEFF = E · Ab = (FR–1) · [EYFP(440)/ECFP(440)], where E is the intrinsic FRET efficiency when fluorophore-tagged molecules are associated with each other, Ab is the fraction of YFP-tagged molecules that are associated with CFP-tagged molecules, and the bracketed term is the ratio of YFP and CFP molar extinction coefficients scaled for the FRET cube excitation filter [30]. We determined this ratio to be 0.094 based on maximal extinction coefficients for ECFP and EYFP [31] and excitation spectra.
Western blot and co-immunoprecipitation
For Western blotting, cells were seeded in 6-well plates. On the next day, 0.5–2 μg/well of cDNA was transfected into cells using the transfection reagent Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. After transfection for 24 h, the cells were harvested as follows. Lysates were prepared in lysis buffer (0.5% Triton X-100, 50 mM HEPES, 120 mM NaCl, 2 mM EDTA, 2 mM MgCl2, pH 7.5 adjusted by NaOH) via passaging 10–15 times through a 26-gauge needle. After lysates were centrifuged at 13,000 × g for 10 min at 4°C, the protein concentration in the supernatants were determined. The extracted proteins in sample buffer were loaded onto 10% Tris-glycine sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) gels. The proteins were transferred onto a nitrocellulose membrane.
For co-immunoprecipitation, 500 μl of cell lysates (500–1,000 μg) were incubated with 1 μg of anti-GFP or anti-Flag antibodies and 30 μl of protein G-agarose beads at 4°C overnight with gentle rotation. After beads were washed three times with wash buffer (same as lysate buffer except for 0.1% Triton X-100 instead of 0.5%), the precipitates were then eluted with 30 μl of 2x Laemmli buffer and subjected to Western blot analysis.
Isometric tension measurement
Isometric tension was measured with a dual-wire multi-channel myograph system (620 M; DMT, Aarhus, Denmark). Mesentery arteries were excised from 11 weeks old male Sprague-Dawley rats and cleaned perivascular adipose tissues in ice-cold NT solution (140 mM NaCl, 5.4 mM KCl, 0.33 mM NaH2PO4, 10 mM HEPES, 10 mM Glucose, 1.8 mM CaCl2 and 1 mM MgCl2 and was of pH 7.4 adjusted with NaOH). Excised arteries were cut into 2 mm arterial ring segments and mounted with 25 μm tungsten wire on NT solution-filled organ chamber for tension recording. For stabilization, mounted arteries were rested on physiological salt solution (118 mM NaCl, 4 mM KCl, 24 mM NaHCO3, 1 mM MgSO4, 0.44 mM NaH2PO4, 5.6 mM glucose and 1.8 mM CaCl2) at least 15 min. with gas mixture (21% O2, 5% CO2, N2 balance) after basal tone of 0.7 g was applied. Whole experiment was maintained with 37°C temperature condition. For the experiment, every arterial segment was conducted with 80 mM KCl-PSS induced contraction for evaluation of vessel integrity and phenylephrine 10 mM was applied to pre-constrict the arteries for further evaluation.
RESULTS
Electrophysiological characteristics of overexpressed Kv7.4 and Kv7.5 channels in HEK293 cells
The electrophysiological characteristics of KCNQ4 (Kv7.4) and KCNQ5 (Kv7.5) channels were evaluated in HEK293 cells. In both Kv7.4- and Kv7.5-expressing HEK293 cells, 1μM of URO-K10 induced robust potassium current (Fig. 2A, F). At +100 mV depolarization, Kv7.4-expressing cells showed 6.08 ± 1.60 nA of whole-cell current while Kv7.5-expressing cells showed 5.26 ± 2.19 nA of whole-cell current. Both channels showed slowly-activating time course upon depolarization, a typical characteristic of KCNQ family. When vehicle solution (control; Normal Tyrode with 1/1,000 (v/v) DMSO) was applied, however, only Kv7.4 channel showed significant potassium current with distinguishable activation time-course. Kv7.5-expressing cells showed nonspecific current amplitude and time courses that are indistinguishable from background current of HEK293 cells. Steady-state current-voltage relationship of both channels showed typical outwardly-rectifying shape (Fig. 2B, G). It is noteworthy to mention that at highly depolarized potentials, 3s pulse duration was rather short to achieve rigorous steady-state in Kv7.5 channels (Fig. 2F). At those potentials, peak currents were used as an alternative to steady-state currents.
Fig. 2
Electrophysiologic characteristics of overexpressed Kv7.4 and Kv7.5 channels in HEK293 cells.
(A) Voltage clamp current traces of control (black) and Kv7.4 with URO-K10 (blue) are shown. (B) Corresponding I–V curve is plotted with steady-state currents measured from –100 mV to +100 mV at 10 mV step intervals. At +100 mV, Kv7.4-expressing cells showed 6.08 ± 1.60 nA whole-cell current in the presence of 1 μM URO-K10. (C) Boltzmann function-fitted conductance curves with URO-K10 show a leftward shift compared to the control (half-maximal voltage of –59.13 ± 6.33 mV). (D) Activation processes are slower with URO-K10 (blue), and (E) activation time constant values increased with the presence of the drug. (F) Voltage clamp traces of control (black) and Kv7.5 with URO-K10 (blue) are shown. (G) I–V curve of Kv7.5 expressing cells is shown. At +100 mV, Kv7.5-expressing cells showed 5.26 ± 2.19 nA whole-cell current in the presence of 1 μM URO-K10. (H) Boltzmann function-fitted steady-state conductance curve of Kv7.5 expressing cells is shown (half-maximal voltage of –74.93 ± 8.64 mV). (I, J) Activation time constant values of Kv7.5-expressing cells in 1 μM URO-K10 are plotted against voltage. In (H) and (J), both regression of conductance-voltage data to Boltzmann function and regression of current-time data to single exponential function were deliberately disregarded in Kv7.5-expressing cells without 1μM URO-K10 (control) (see manuscript for detailed description).
![]()
Voltage-dependency of activation processes of two channels were further analyzed from conductance-voltage (G–V) curves. Steady-state conductances from 1 μM URO-K10 activated currents fitted well with Boltzmann function (Fig. 2C, H). Half-maximal voltage of URO-K10 activated Kv7.4 showed –59.13 ± 6.33 mV while Kv7.5 showed –74.93 ± 8.64 mV. Likewise, steady-state conductances from intrinsic Kv7.4 current (control) fitted well with Boltzmann function, yielding half-maximal voltage of –35.58 ± 4.49 mV. Since Kv7.5 channels did not show significant intrinsic activity, we deliberately disregarded Kv7.5 channels from G-V analysis. In a sense, steady-state current-voltage relationship of intrinsic Kv7.5 current merely showed an ohmic curve (Fig. 2G).
URO-K10 (1 μM) not only potently increased whole-cell current of both Kv7.4 and Kv7.5 channels but also significantly prolongated the activation process (Fig. 2D, E, I, J). Activation time courses of Kv7.4 channel at 5 different membrane potentials (–40, –20, 0, 20, 40 mV) were fitted into single exponential activation function. Mathematical formulation for time-dependency of conductance was same as Hodgkin-Huxley model of delayed rectifier K channels [27]. However, the order of exponential component was set to 1 since higher magnitudes were not suitable for remarkably slow activation process of KCNQ family. Nevertheless, 1 μM URO-K10 activated Kv7.4 channels showed activation time constants of 0.38 ± 0.06 sec (–40 mV), 0.32 ± 0.07 sec (–20 mV), 0.30 ± 0.08 sec (0 mV), 0.31 ± 0.08 sec (20 mV), and 0.32 ± 0.08 sec (40 mV), while time constants of intrinsic activity (control) showed 0.15 ± 0.04 sec (–40 mV), 0.13 ± 0.04 sec (–20 mV), 0.10 ± 0.02 sec (0 mV), 0.08 ± 0.02 sec (20 mV), and 0.08 ± 0.02 sec (40 mV). Prolongation of activation processes by URO-K10 was also observed in Kv7.5 channels, however, a single exponential component was not enough for successful description of the activation process. Rather, the URO-K10 activated current time courses were fitted well with double exponential components, hence yielding two independent time constants (Fig. 2I, J). Slow time constants (τslow) of Kv7.5 channels showed 0.70 ± 0.16 (–40 mV), 0.73 ± 0.21 sec (–20 mV), 0.83 ± 0.26 sec (0 mV), 0.95 ± 0.32 sec (20 mV), and 1.05 ± 0.30 sec (40 mV), respectively. Fast time constants (τfast) of the channels showed 0.09 ± 0.04 (–40 mV), 0.05 ± 0.02 sec (–20 mV), 0.04 ± 0.02 sec (0 mV), 0.03 ± 0.01 sec (20 mV), and 0.03 ± 0.0.02 sec (40 mV), respectively.
The results suggest that 1 μM URO-K10 potently activates Kv7.4 and Kv7.5 channels, and that Kv7.5 channels do not permit any significant channel-specific potassium current unless pharmacological agonists facilitate the channel. URO-K10 (1 μM) not only increased whole-cell current of the channels but also significantly prolongated activation processes.
Dose-dependent action of URO-K10 onto Kv7.4 and Kv7.5
To acquire EC50 values of URO-K10, dose-dependent currents were measured using a 10 second voltage step at +50 mV (Fig. 3A, C). EC50 values were evaluated to be 210.8 nM and 142.0 nM in Kv7.4 and Kv7.5 respectively. Application of increasing concentrations of URO-K10 produced increases in the current amplitude of Kv7.4 (Fig. 3B). As a remark, in Kv7.4-expressing cells, c-type inactivation [32] was observed in concentrations larger than 1 μM. However, as Kv7.5 channels have been known to get suppressed in higher doses of agonists according to recent results [33], a similar reduction was observed in concentrations larger than 3 μM (Fig. 3D).
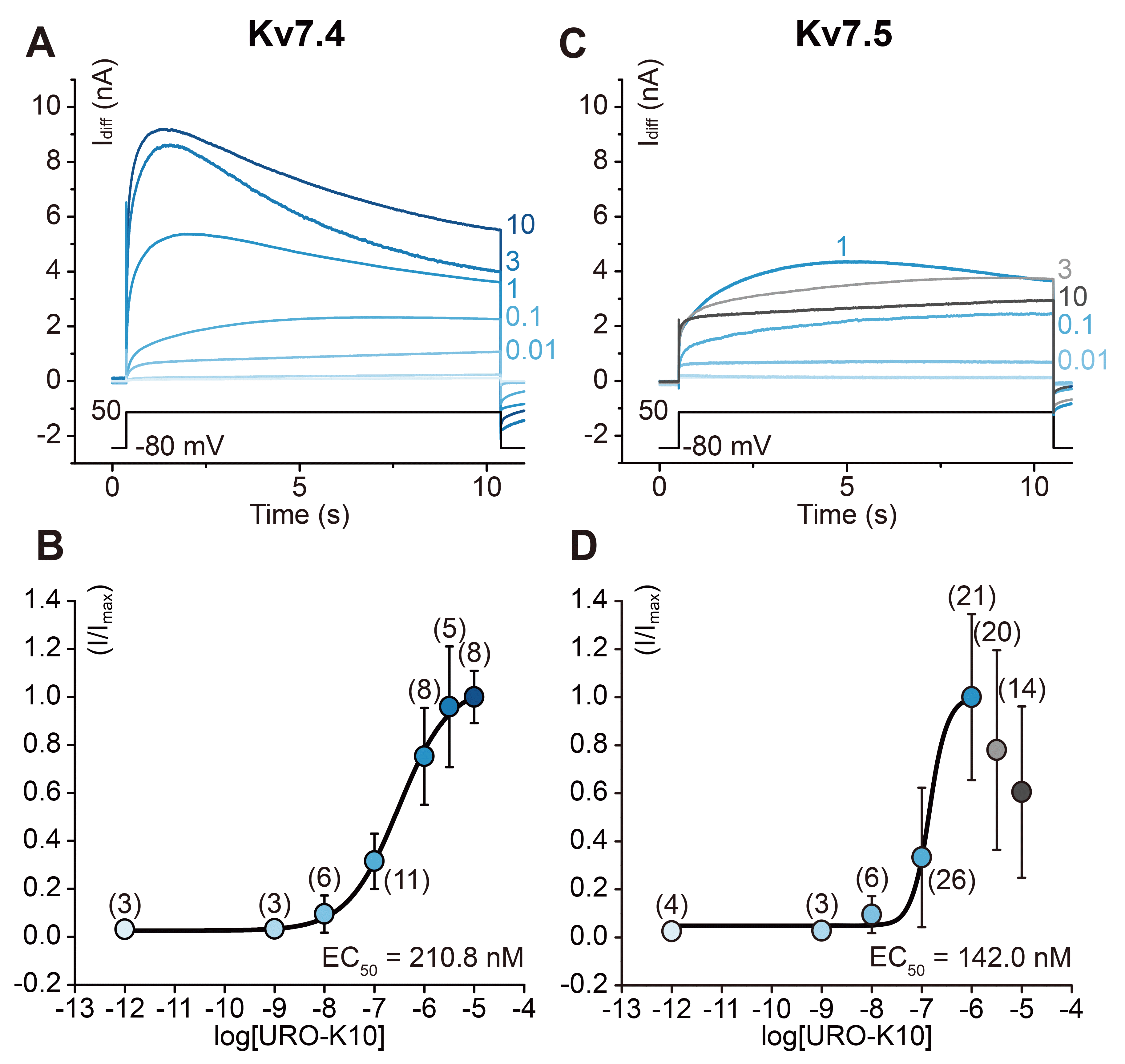
Fig. 3
Dose-dependent action of URO-K10 on Kv7.4 and Kv7.5.
(A) Kv7.4 current traces are obtained at +50 mV with URO-K10 concentrations ranging from 1 pM to 10 μM. At each concentration, drug-affected current traces at +50 mV was subtracted from drug-free current traces at the same cell and at the same membrane potential (Idiff). (B) Dose-response curve in a logarithmic scale shows EC50 value of 210.8 nM for Kv7.4. (C, D) Dose-dependent action of URO-K10 on Kv7.5 is also shown in parallel. Dose-response curve in a logarithmic scale shows EC50 value of 142.0 nM for Kv7.5. In both (A) and (C), black inset lines indicate corresponding voltage-clamp step.
![]()
URO-K10 increases both open probability and single channel conductance of Kv7.4 channel
As a next step, we examined the effect of URO-K10 in a single-channel level. In HEK293 cells expressing KCNQ4 channels, we measured single-channel currents of the channel in a cell-attached configuration since conservation of physiological intracellular milieu such as calmodulin [5,8,34], PIP2 [23,35-38] are essential for KCNQ4 channels to open. As a result, 1 μM URO-K10 not only increased open probability of the channel but also increased slope conductances (Fig. 4A). Mean open probability of (Po) KCNQ4 in 30 sec recording interval without URO-K10 was 0.15 while 1 μM URO-K10 increased the Po up to 0.46 (Fig. 4B). Slope conductances from I–140 mV–I–200 mV was 4.77pS for intrinsic KCNQ4 activity and 18.85pS when 1μM URO-K10 was applied (Fig. 4C).

Fig. 4
URO-K10 increases both open probability and slope conductance of KCNQ4 channel.
(A) Current traces from cell-attached single channel recordings at given commanding potentials. Traces in blue lines indicate pipette solution with URO-K10 (1 μM) while traces in black lines indicate control. At each commanding potential, dotted lines and full lines indicate closed states (C) and open states (O), respectively. (B) Open probabilities of KCNQ4 channel in 30 sec recording interval. In both traces, –160 mV of commanding potential was applied. URO-K10 (1 μM) significantly increased mean open probability of KCNQ4 channel (0.15 vs. 0.46). (C) Slope conductances calculated from unitary current amplitude at given commanding potential. 1 μM URO-K10 significantly increased slope conductance of KCNQ4 channel (4.77 pS vs. 18.85 pS).
![]()
Kv7.4 and Kv7.5 have a molecular interaction with ancillary subunit KCNE4
The ancillary subunit KCNE4 by itself cannot form a channel pore, but has been known to modulate the expression and activity of channels formed by KCNQ subunits. KCNE4 is expressed along with Kv7.4 in vasculatures, particularly in the mesenteric artery [17] and in corpus cavernosum smooth muscle cells [14]. In our hands, the results showed that when expressed in HEK293 cells, Kv7.4, Kv7.5 and KCNE4 indeed co-localized within the cell. FRET analysis using CFP-tagged Kv7.4, Kv7.5 and YFP-tagged KCNE4 was performed, and regions with increased FRET efficiency were indicated by intensity gradient (Fig. 5B). Compared to the controls, CFP-tagged Kv7.4, Kv7.5 and YFP-tagged KCNE4 showed increased FRET efficiency (11.3% ± 1.5% for KCNQ4-ECFP/KCNE4-EYFP pair, and 12.5% ± 1.6% for KCNQ5-ECFP/KCNE4-EYFP pair, respectively). Interestingly, KCNE4 localization in the plasma membrane was prominent only when it was co-expressed with Kv7.4 or Kv7.5 (Fig. 5A, B), a possible indication of the limited membrane trafficking properties of the ancillary subunit. Line-scan analysis clearly shows that YFP signal from KCNE4 subunit (KCNE4-EYFP) is limited in cytosolic level and no significant signal is found near plasma membrane borderline (light gray lines, Fig. 5A, B). Co-expression of KCNQ4 or KCNQ5 translocated the KCNE4-YFP signal near plasma membrane and high FRET signal was observed near plasma membrane, simultaneously. Further in vitro analysis of protein-protein interaction between KCNQ4, KCNQ5 and KCNE4 was performed via co-IP analysis. Flag-tagged KCNE4 was pulled down along with CFP-tagged KCNQ4 and KCNQ5 and vice versa, indicating tight molecular interaction between the two (Fig. 5C). These results suggest that KCNE4 interacts with both KCNQ4 and KCNQ5, and that plasma membrane localization of KCNE4 is greatly enhanced when pore-forming subunits (KCNQ4, KCNQ5) are co-expressed.

Fig. 5
Kv7.4 and Kv7.5 have a molecular interaction with ancillary subunit KCNE4.
(A) Fluorescence imaging of KCNQ4-ECFP, KCNQ5-ECFP and KCNE4-EYFP. Both KCNQ4-ECFP and KCNQ5-ECFP showed strong cyan fluorescence in plasma membrane and inside the cell. KCNE4-EYFP, however, showed discrete, puncta-like YFP signal inside the cell while signal in plasma membrane was highly limited. (B) Fluorescence imaging of KCNQ4-ECFP+KCNE4-EYFP and KCNQ5-ECFP+KCNE4-EYFP. When co-expressed with KCNQ4 or KCNQ5, prominent localization of YFP signal in plasma membrane was observable. High effective FRET efficiency (EEFF, see Methods) between KCNQ4-ECFP and KCNE4-EYFP (11.3% ± 1.5%, n = 7), and between KCNQ5-ECFP and KCNE4-EYFP (12.5% ± 1.6%, n = 7) were also observable, suggesting strong in vitro interaction between each α subunit and KCNE4. (C) Co-immunoprecipitation blot shows interaction with KCNE4 at a molecular level. KCNQ4-EGFP band (104 kDa) and KCNQ5-EGFP band (130 kDa) is observed when pulled down with the anti-Flag antibody. Conversely, KCNE4-Flag (23 kDa) is observed when pulled down with anti-GFP.
![]()
Electrophysiology of KCNQ4/KCNE4 and KCNQ5/KCNE4 complex
Since protein-protein interaction between KCNE4 and KCNQ4, KCNQ5 have been verified, we investigated whether expression of KCNE4 alters electrophysiological property of KCNQ4 and KCNQ5 channels. As a result, expression of KCNE4 suppressed intrinsic activity of KCNQ4 (Fig. 6A, G). Expression of KCNE4 had little effect to intrinsic KCNQ5 activity; KCNQ5/KCNE4 expressing HEK293 cells showed nonspecific background current (Fig. 6D, G). URO-K10 (1 μM) increased whole-cell potassium currents in both KCNQ4/KCNE4 expressing cells (1.66 ± 0.72 nA, 100 mV) and KCNQ5/KCNE4 expressing cells (2.51 ± 1.29 nA, 100 mV). Boltzmann fitting of 1 μM URO-K10 activated KCNQ4/KCNE4 channels and KCNQ5/KCNE4 channels showed that the half-maximal voltage of the former is –67.35 ± 5.50 mV and the latter is –72.73 ± 7.09 mV (Fig. 6C, F). As previously mentioned, whole-cell Kv7.5 current without URO-K10 was disregarded from voltage-dependency analysis. The overall effect of URO-K10 and KCNE4 onto KCNQ4 and KCNQ5 was summarized with respect to maximum steady-state conductance and half-maximal voltage (Fig. 6G, H). First, 1 μM URO-K10 potently increased maximum steady-state conductance of both KCNQ4 and KCNQ5 channels. This effect, however, was suppressed when KCNE4 was co-expressed with KCNQ4 or KCNQ5 channels. Therefore, the results suggest that KCNE4 inhibits agonist-activated KCNQ4 or KCNQ5 current through strong interaction. In terms of half-maximal voltage, 1 μM URO-K10 shifted the activation curve towards hyperpolarized potentials. Interestingly, KCNE4 seemed to exert little or no effect to voltage-dependency of the activation process of the KCNQ4 channel as V1/2Q4 (–35.58 ± 4.49 mV, n = 4) and V1/2Q4E4 (–29.17 ± 13.27 mV, n = 9) are statistically indifferent as well as URO-K10 activated V1/2Q4U (–59.13 ± 6.33 mV, n = 11) and V1/2Q4E4U (–67.35 ± 5.50 mV, n = 12) are similar.

Fig. 6
Electrophysiology of KCNQ4/KCNE4 and KCNQ5/KCNE4 complex.
(A) KCNE4 suppresses the activity of KCNQ4, as shown in the whole-cell potassium current traces. Whole-cell potassium current in KCNQ4/KCNE4 cells is 1.66 ± 0.72 nA at +100 mV. (B, C) I–V and conductance (G–V) curves are shown for control (KCNQ4/E4, black) and with 1 μM URO-K10 (blue). Half-maximal voltage in the presence of the drug is –67.35 ± 5.50 mV. (D–F) Electrophysiological characteristics of KCNQ5/KCNE4 are also shown in parallel. In 1 μM URO-K10, whole cell potassium current in KCNQ5/KCNE4 was 2.51 ± 1.29 nA at +100 mV and the half-maximal voltage was –72.73 ± 7.09 mV. (G) Maximum steady-state conductances of KCNQ4/E4 and KCNQ5/E4 are compared. KCNE4 suppresses both agonist-activated and intrinsic current of KCNQ4. KCNE4 suppresses the agonist-activated current of KCNQ5, but its effects on intrinsic currents are not shown due to the small current size indistinguishable from the background HEK293 cell current. (H) Half maximal voltages of KCNQ4/E4 and KCNQ5/E4 are compared. KCNE4 exerts a negligible effect on the voltage-dependency of KCNQ4 and KCNQ5 channels. **p < 0.05.
![]()
Effect of URO-K10 onto mesenteric arterial vasorelaxation
Among 5 KCNE subunits (KCNE1–KCNE5), it was reported that transcription and expression of KCNE4 is the most abundant in mesenteric vascular smooth muscle cells [17,19,25,26]. Moreover, the expression of KCNQ4 was most abundant in mesenteric vascular smooth muscle cells among 5 KCNQ subunits. Therefore, we tested if URO-K10 could induce vascular relaxation. As a result, 1 μM of URO-K10 almost completely relaxed 10uM phenylephrine-induced contraction (11.42% ± 2.06%, n = 4) (Fig. 7A, C). When extracellular potassium concentration was raised up to 80 mM, however, little or only partial relaxation was observed (86.88% ± 3.05%, n = 6) (Fig. 7B, C). These results suggest that URO-K10 mediated relaxation is highly sensitive to potassium concentration gradient, hence via URO-K10-sensitive potassium channels. In order to measure EC50 in a tissue-specific scale, 0.1 μM to 1 μM of URO-K10 was applied in a serial manner. As a result, EC50 was estimated as 521.0 nM (Fig. 7D, E).
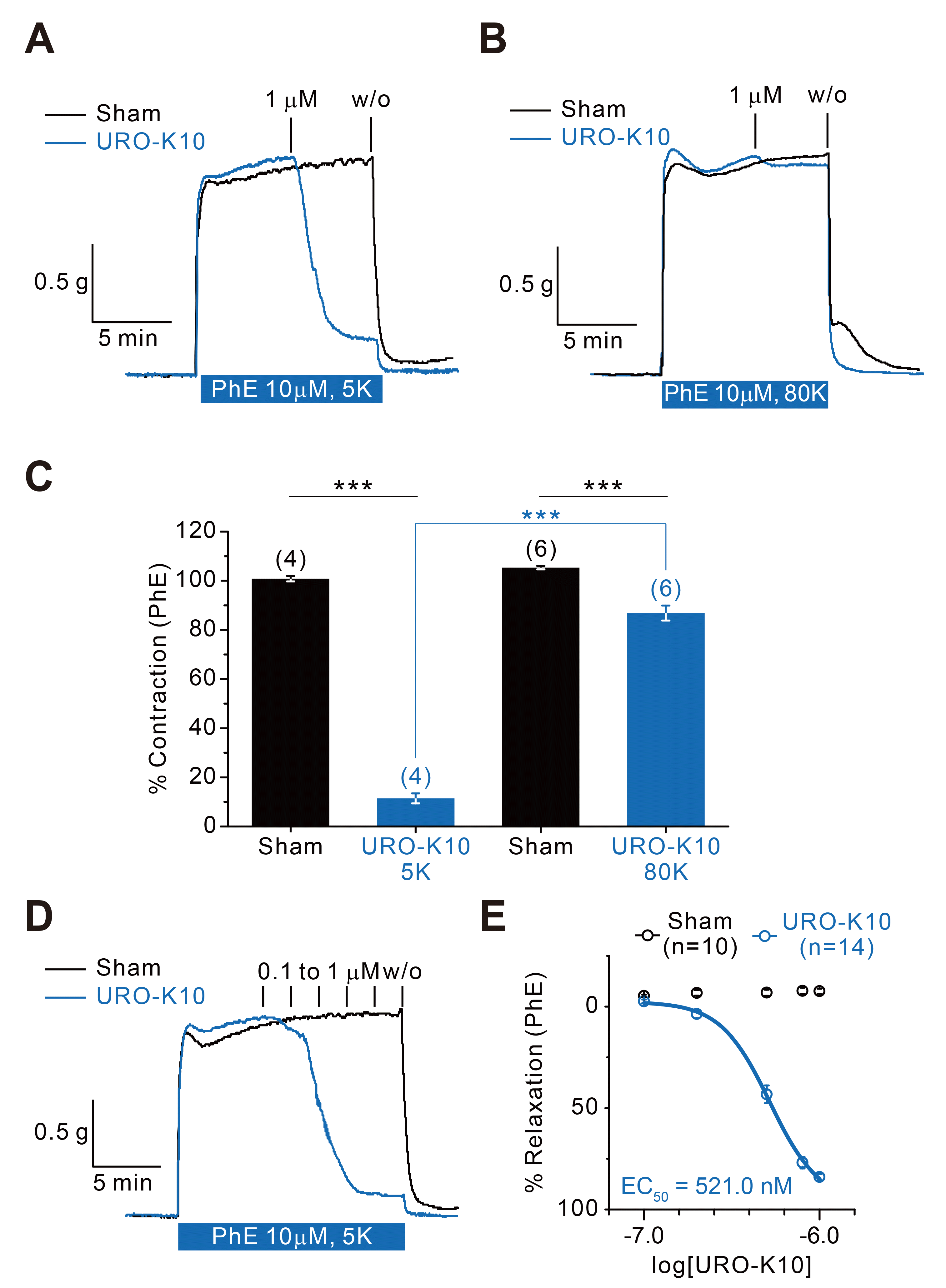
Fig. 7
Effect of URO-K10 onto mesenteric arterial vasorelaxation.
(A) Isometric pressure myograph shows almost complete relaxation of phenylephrine-induced contraction upon administration of 1 μM URO-K10 (blue) (11.42% ± 2.06%, n = 4). (B) This relaxation effect is abolished in 80 mM potassium extracellular concentration (86.88% ± 3.05%, n = 6). (C) Bar graph summarizes percentages of contraction with and without 1 μM URO-K10 in 5 mM or 80 mM potassium. (D) Various concentrations of URO-K10 (from 0.1 to 1 μM) was applied to measure EC50 in a tissue-specific scale. (E) Percentages of contraction is plotted against concentration at a logarithmic scale. EC50 is estimated to be 521.0 nM. ***p < 0.001.
![]()
DISCUSSION
In our hands, both KCNQ4 and KCNQ5 channels expressed in HEK293 cells showed robust facilitation when 1 μM of URO-K10 was applied (Fig. 2A–H). Moreover, 1 μM URO-K10 increased the currents evoked by potentials higher than –80 mV, hence steady-state conductances. At the same time, 1 μM URO-K10 also prolongated activation processes at each voltage steps (Fig. 2D–J). Although KCNQ4 channels showed intrinsic activity without URO-K10, KCNQ5 channels showed no significant intrinsic activities. In other words, KCNQ5-expressing cells showed only nonspecific, background-like whole-cell currents at each voltage steps when control (Normal Tyrode with DMSO) external solution was applied. The absence of intrinsic activity of KCNQ5 observed in this study, however, is somewhat different from previous reports. KCNQ5-specific whole-cell current from KCNQ5-expressing A7R5 smooth muscle cell lines showed that KCNQ5 channels possess significant intrinsic activity [33]. Similar results were also observed in Xenopus Oocyte expression system [22,39]. Interestingly, the same cDNA construct of KCNQ5 elicited highly limited amount of KCNQ-like potassium current when transfected in HEK293 cells [22]. Although it is difficult to dissect the source of such discrepancy, the difference may have been originated from intrinsic characteristics of each expression system and means of introducing heterologous cDNA sequences or mature mRNA transcript of KCNQ5. Based on the dynamic responses of KCNQ1 channel by intracellular conditions such as intracellular calcium concentration and binding of calmodulin [5,8,34], binding of phosphatidylinositol-4,5-bisphosphate (PIP2) [23,35,37], and others, the intrinsic activity of KCNQ5 may differ from cell to cell in terms of variations in intracellular determinants.
The pharmacological dose-response curve showed that EC50 of URO-K10 onto KCNQ4 is 210.8 nM. KCNQ5 showed higher sensitivity to the drug: EC50 = 142.0 nM. Interestingly, URO-K10 showed partial antagonistic effect onto KCNQ5 on doses higher than 3 μM (Fig. 3C, D; light gray and gray curves). This partial antagonistic effect can also be seen in other KCNQ agonists such as ML-213 and ICA-069673 [33]. Considering stark differences in molecular structures of each agonist (Fig. 1), partial antagonistic effect may seldom be attributable to singular allosteric site within the channel. In a sense, the chemical structure of URO-K10 strongly negates the possibility of binding site being similar to ML-213 or ICA-069673. Provided, it is intriguing enough that 10 μM of XE-991, a KCNQ blocker, inhibited the agonistic action of both URO-K10 and ML-213 (Supplementary Fig. 1).
In a single channel level, 1 μM URO-K10 not only increased open probability of KCNQ4 channel but also increased slope conductances (Fig. 4A–C). Since the measurement was done in cell-attached configuration, it is impossible to calculate absolute conductance of the channel at given commanding voltage (Vcmd) unless the resting membrane potential at certain condition is known (Vmem = Vres – Vcmd). If we surmise the resting potential of KCNQ4-expressing HEK293 cells without URO-K10 to be around –60 mV and the resting potential of KCNQ4-expressing HEK293 cells with 1 μM URO-K10 to be around –80 mV based on current-clamp results (Supplementary Fig. 2), –160 mV of commanding potential would correspond to 100 mV of transmembrane potential (Vmem) and 80 mV, respectively. The calculation leads to single channel conductance of 2.69 pS for intrinsic KCNQ4 activity, which is highly similar to previously reported value (2.1 pS) [40]. Moreover, the same calculation yields 7.70 pS for 1 μM URO-K10 activated KCNQ4 activity. Overall effect of the drug would therefore be 2.86 (single channel conductance) × 3.09 (open probability) = 8.84 times increase in whole-cell conductance provided that number of the channels in plasma membrane is constant. The calculation based on single-channel recording, however, seems to partially overestimate the effect of the drug since the actual whole-cell recording shows slightly smaller increase than the calculated value (Fig. 6G).
As mentioned earlier, the expression and interaction of KCNE4 and KCNQ4 in mesenteric vascular smooth muscle has already been addressed. In our hands, phenylephrine-induced vasoconstriction was almost fully recovered by 1 μM URO-K10. The relaxation was highly depending on potassium concentration gradient (Fig. 7A–C). Therefore, URO-K10 mediated vasorelaxation would possibly be due to shift of membrane potential towards Nernst equilibrium potential of potassium ions at given potassium concentration gradient. Nevertheless, EC50 of URO-K10 on tissue level was 521.0 nM, a higher value than the one measured in cellular level.
FRET analysis among KCNQ4-ECFP, KCNQ5-ECFP and KCNE4-EYFP strongly suggested that interaction between KCNQ4-KCNE4 and KCNQ5-KCNE4 are significant. Interes­tingly, it was evident that YFP signal from KCNE4-EYFP was highly confined in intracellular area when only KCNE4-EYFP is expressed without any of α subunits (Fig. 5A). However, we do not know yet the exact intracellular compartment of which KCNE4 subunits reside. It is also important to remark that somewhat aggregated, puncta-like localization of YFP signals inside the cell were prominently seen in KCNE4-EYFP expressing cells. Confocal microscopy may have been helpful to dissect the exact location of the signal. Although it is highly unlikely, puncta-like signal inside the cell from KCNE4-EYFP might have been originated from superior or inferior plasma membrane; not a single puncta-like signal was observable in lateral membrane from all imaging sessions (n = 7). Puncta-like distribution inside the cell may also indicate putative localization of KCNE4 inside intracellular organelles such as ER-Golgi system, endosome, and others. The intracellular, puncta-like distribution of KCNE4 was rescued by co-expression of KCNQ4, KCNQ5 α subunits (Fig. 5A, B). In our hands, the stark difference in intracellular localization of KCNE4 depending on co-expression of α subunits was clear enough that even line-scan analysis showed clear distinction. The once puncta-like distribution of KCNE4 was completely changed as soon as α subunits are co-expressed and localized inside the plasma membrane. It is intriguing, however, that the endogenous distribution of KCNQ4/KCNQ5 or KCNQ4/KCNE4 complex seem to follow the puncta-like localization [14,17]. In isolated corpus cavernosum smooth muscle cells, proximal ligation assay (PLA) using anti-KCNQ4 Ab and anti-KCNQ5 Ab showed puncta-like signals and in isolated mesenteric arterial smooth muscle cells, PLA using anti-KCNQ4 Ab and anti-KCNE4 Ab showed similar puncta-like signals inside the cell. Besides to FRET analysis, co-IP results also support the strong interaction between KCNE4 and KCNQ4, 5 subunits (Fig. 5C). Moreover, the functional consequences of such interaction were shown to be inhibitory in terms of potassium current carried by α subunits in each complex (Fig. 6). Although maximum steady-conductances were severely reduced by KCNE4 subunit, the interaction of KCNE4 seems to exert little or no effect to voltage-dependency of activation process for half-maximal voltage rarely varied.
According to Seefeld et al. [24], the supremacy of the URO-K10’s motif compound was enormous compared to other previously known KCNQ activators (ML-213 and ICA-27243). For instance, EC50 of the motif compound was 80 times lower than ML-213, and 500 times lower than ICA-27243 [24]. In our hands, increase in steady-state conductance at +50 mV was significantly higher in 1 μM URO-K10 applied cells than in 1 μM ML-213 applied cells (Supplementary Fig. 3). In a sense, 1 μM of URO-K10 evoked KCNQ4, 5 current even near –80 mV, a more hyperpolarized potential than resting membrane potential of most smooth muscle cells. This would be largely attributable to left-shift of activation curves of KCNQ4 and 5 by URO-K10. Therefore, we expect that micromolar concentration of URO-K10 may induce significant potassium efflux and consequent membrane hyperpolarization in smooth muscle cells even in resting state, provided that expression level of KCNQ4 or 5 channels in the cell is sufficient enough. The hyperpolarization of resting membrane potential would require stronger depolarization for sufficient L-type calcium channel activation to occur. Even if the increase of steady-state conductance by URO-K10 is paramount, however, one must not overlook the other effect of URO-K10: prolongation of activation process. According to our results (Fig. 2E, J) activation time constants of KCNQ4 and KCNQ5 was in range of 0.25–0.4 sec and 0.7–1.1 sec, respectively, in –40 mV–40 mV membrane potential. If we approximate the mean time constant of two channels be 0.4 sec in those potential range, 0.28 sec, 0.92 sec and 1.84 sec is required for 50%, 90% and 99% of maximum pharmacological effect at given dose be achieved. Such a slow activation may clearly dissect both pros and cons of the drug according to target tissue or purpose of the drug. In vascular smooth muscle cells, for instance, continuity of membrane potential of the cell without any action potential (AP) firing may guarantee successful action of the drug. On the other hand, the efficacy of the drug in AP firing smooth muscle cells may not be efficient as well. For instance, both periodic wave and occasional spikes can be seen along various time scales in GI smooth muscles. Considering the longest time scale, for example, 3 sec as in slow waves, the time interval is barely sufficient for complete action of the drug. It is also noteworthy to remark that the calculations of both time constants and subsequent time estimates for corresponding percentage of the drug action have been done in potential range from the minimum depolarization threshold for L-type calcium channel activation (–40 mV) to maximum peak potential of the spikes in some of the AP-firing smooth muscle cells (+40 mV).
Given that genuine purpose of URO-K10 was to elicit as large potassium current across the membrane as possible through KCNQ4, 5 channels, the co-existence of KCNE4 in the membrane may be seen as one of the major obstacles. In other words, the response would be more effective in tissues where only KCNQs are expressed and no or little KCNE. Yet, almost complete relaxation of mesenteric arterial vasoconstriction by URO-K10 assures similar promising effect on corpus cavernosum where expression of both KCNQ4 and KCNE4 are as significant as mesenteric vascular smooth muscle cells. A nonspecific vasodilation may well lead to detrimental hypotension of which all potassium channel activators intrinsically possess as a major adverse effect. In a sense, strong vasodilative effect of URO-K10 may provoke the adverse effect, however, it shall be noted that blood pressure at rest is largely determined by collective systemic resistance of arterioles, especially arterioles within skeletal muscle. Therefore, vasodilative effect of URO-K10 must be addressed in skeletal muscle arterioles as well as in corpus cavernosum smooth muscle per se. Dose-dependency in both tissues may result in EC50s, hence putative therapeutic window in tissue level.
Aside from pharmacodynamic variables of URO-K10, tonic contractility of corpus cavernosum ensures some promising results. Recall that 1 μM URO-K10 almost completely relaxed phenylephrine-induced mesenteric artery vasoconstriction, however, the relaxation could not be extended over basal contraction tone of the muscle strip (Fig. 7). In other words, the drug action was supreme only if there had been a priori contraction. Therefore, it is yet uncertain whether URO-K10 may exert vasodilative effect on rather flaccid smooth muscle strips. Since corpus cavernosum is one of the rare smooth muscles whose resting contraction tones are high, this tonic contractility may serve as a good substrate for drug to intervene. In this context, prospective studies in involuntary sphincter muscles with URO-K10 seem to be both intriguing and necessary.
SUPPLEMENTARY MATERIALS
Supplementary data including three figures can be found with this article online at http://pdf.medrang.co.kr/paper/pdf/Kjpp/Kjpp2020-24-06-07-s001.pdf.
 XML Download
XML Download