

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
METHODS
Short-term in vivo T3 treatment
Assessment of the hyperthyroid state
Tension measurements
Acute in vitro T3 treatment
Cumulative concentration-response curves (CRCs) for T3 in aortic tissues pre-contracted with PHE
CRCs to relaxant and contractile agonists in in vitro T3-treated aortic tissues
Short-term in vivo T3 treatment
CRCs to relaxant agonists in aortic tissues from short-term in vivo T3-treated rats
Contractile stimuli in aortic tissues of short-term in vivo T3-treated rats
Chemicals
Statistical analysis
RESULTS
PHE-induced pre-contractions in aortic tissues are not altered by T3 in vitro application
 | Fig. 1Triiodothyronine (T3) applied in vitro did not influence previously established aortic active tonus.Cumulative administration of increasing concentrations of T3, every 30 min, in endothelium intact (Endo +) aortic rings contracted with phenylephrine (1 μM). Temporal controls received the corresponding concentration of vehicle (V). Values are presented as means ± standard error of the means (n = 16). p = not significant (two-way ANOVA).
|
Relaxant and contractile responses to selective agonists in aortic tissues stayed unchanged by in vitro T3-treatments
 | Fig. 2Lack of effect of 3,5,3’ triiodo-L-thyronine (T3) in vitro treatments on the relaxing responses of agonists in aortic tissues.(A, B) Relaxing responses to acetylcholine and (C, D) sodium nitroprusside (SNP) in endothelium-intact aortic rings (Endo +) contracted with phenylephrine (1 μM), treated previously with T3 (0.01, 0.1, and 1 μM) or vehicle (V) during 30 min or 2 h. Vehicle concentration corresponds to that required to dissolve T3 1 μM. Relaxing responses were calculated as decreases of phenylephrine-induced tension (grams). Values are presented as means ± standard error of the means (n = 15–16). p = not significant (two-way ANOVA).
|
 | Fig. 3Lack of effect of in vitro 3,5,3’ triiodo-L-thyronine (T3) treatments on the contractile responses to agonists in aortic segments.(A, B) Cumulative concentration-response curves to Phenylephrine and (C, D) Angiotensin II in annular segments of aortas with endothelium (Endo +) treated previously with T3 (0.01, 0.1, and 1 μM) or vehicle (V) for 30 min and 2 h. Vehicle concentration corresponded to that required to dissolve T3 1 μM. The contractile responses represent grams of developed tension. Values are presented as means ± standard error of the means (n = 16). p = not significant (two-way ANOVA).
|
Short-term in vivo T3-treatment efficiently induced hyperthyroidism in rats
Table 1
| Treatment | fT3 (pg/ml) | TSH (ng/ml) | BM (g) | LVdM/LT (mg/mm) |
|---|---|---|---|---|
| V1d | 2.54 ± 0.18 | 1.79 ± 0.18 | 286.82 ± 3.52 | 4.16 ± 0.24 |
| T31d | 6.38 ± 0.49a | 0.28 ± 0.07a | 282.74 ± 3.57 | 4.69 ± 0.33 |
| V3d | 2.11 ± 0.27 | 1.83 ± 0.11 | 292.45 ± 3.75 | 4.33 ± 0.30 |
| T33d | 9.91 ± 0.72b,c | 0.18 ± 0.02b | 275.56 ± 3.60d | 5.66 ± 0.25e |
Values are presented as means ± standard error of the means. Serum levels of free 3,5,3’ triiodo-L-thyronine (fT3) and thyroid stimulating hormone (TSH). Body mass (BM) values. Left ventricular dry mass to tibia length (LVdM/TL) ratio as an indicator of cardiac hypertrophy. Short-term hyperthyroidism in rats was induced by daily subcutaneous injection of T3 (500 μg·kg–1) for 1 (T31d) and 3 (T33d) days. Control animals were injected every day with the vehicle (1 ml·kg–1·d–1) for 1 (V1d) and 3 (V3d) days. ap < 0.05 vs. V1d, bp < 0.05 vs. V3d, cp < 0.05 vs. T31d, n = 6 rats per group (one-way ANOVA with Tukey post-hoc test). dp < 0.05 vs. V3d, n = 30 rats per group (unpaired Student t-test). ep < 0.05 vs. V3d; n = 8 hearts per group (one-way ANOVA with Tukey post-hoc test).
![]()
Relaxant responses to selective agonists remained unaltered in aortic tissues of short-term in vivo T3-treated rats
 | Fig. 4Short-term in vivo 3,5,3’ triiodo-L-thyronine (T3) treatment did not affect relaxant responses of agonists in aortic tissues.(A) Cumulative concentration-relaxing response curves to acetylcholine and (B) sodium nitroprusside (SNP) in endothelium-intact (Endo +) thoracic aortic rings isolated from rats treated subcutaneously with T3 (500 μg·kg–1·d–1) or vehicle (V; 1 ml·kg–1·d–1) for 1 and 3 days (T31d, T33d and V1d, V3d, respectively). Relaxing responses were calculated as decreases of phenylephrine-induced tension. Values are presented as means ± standard error of the means (n = 14–16). p = not significant (two-way ANOVA).
|
Depression of contractile responses to PHE, ANG II, and high K+ in aortic tissues from short-term in vivo T3-treated rats
 | Fig. 5Delayed depression of Phenylephrine-induced contractions in aortic tissues from short-term thyroid hormone-treated rats.Cumulative concentration-contractile response curves to phenylephrine, in endothelium-intact (Endo +) thoracic aortic rings isolated from rats treated subcutaneously with 3,5,3’ triiodo-L-thyronine (T3; 500 μg·kg–1·d–1) or vehicle (V; 1 ml·kg–1·d–1) for 1 and 3 days (T31d, T33d and V1d, V3d, respectively). Contractile responses are expressed as grams (g) of developed force. Values are presented as means ± SEM (n = 16). *p < 0.05 T33d vs. V3d (two-way ANOVA with Sidak’s post-hoc test).
|
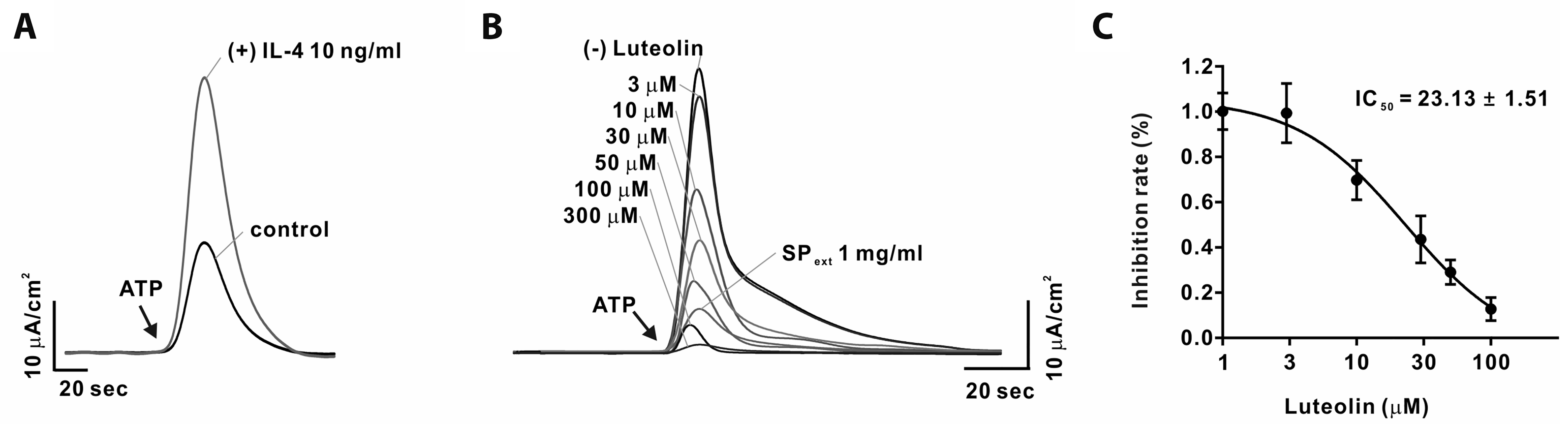 | Fig. 6Endothelium-independent inhibition of contractile responses to Phenylephrine in aortic tissues from 3,5,3’ triiodo-L-thyronine (T3) in vivo treated rats.(A) Cumulative concentration-response curves (CRCs) to phenylephrine in annular segments of aortas with (Endo +) and without (Endo –) endothelium. (B) CRCs to phenylephrine in endothelium-intact (Endo +) thoracic aortic rings in the presence or absence of NG-Nitro-L-arginine methyl ester (L-NAME, 100 μM). Aortic tissues were obtained from rats treated with subcutaneous injections of T3 (500 μg·kg–1·d–1) or vehicle (1 ml·kg–1·d–1) for 3 days (T33d and V3d, respectively). All data are expressed as grams (g) of developed force. Values are presented as means ± standard error of the means. (A) (n = 16) *p < 0.05 T33d (Endo +) vs. V3d (Endo +); #p < 0.05 T33d (Endo –) vs. V3d (Endo –); ψp < 0.05 T33d (Endo +) vs. T33d (Endo –); &p < 0.05 V3d (Endo +) vs. V3d (Endo –). Two-way ANOVA with Sidak’s post-hoc test. (B) (n = 14–16) *p < 0.05 T33d vs. V3d; #p < 0.05 T33d L-NAME vs. V3d L-NAME; ψp < 0.05 T33d vs. T33d L-NAME; &p < 0.05 V3d vs. V3d L-NAME. Two-way ANOVA with Sidak’s post-hoc test.
|
 | Fig. 7Endothelium-independent inhibition of contractile responses to angiotensin II and high K+ in aortic tissues from thyroid hormone-treated rats.(A) Cumulative concentration-response curves to angiotensin II in endothelium-denuded (Endo –) thoracic aortic rings. (B) Contractile responses to high K+ (40 and 80 mM) in endothelium-denuded (Endo –) thoracic aortic rings. Aortic tissues were obtained from rats treated with subcutaneous injections of 3,5,3’ triiodo-L-thyronine (500 μg·kg–1·d–1) or vehicle (1 ml·kg–1·d–1) for 3 days (T33d and V3d, respectively). All data are expressed as grams (g) of developed force. Values are presented as means ± standard error of the means. (A) (n = 15) *p < 0.05 vs. V3d (two-way ANOVA with Sidak’s post-hoc test). (B) (n = 13–15) *p < 0.05 vs. V3d (K+ 40 mM); #p < 0.05 vs. V3d (K+ 80 mM) (unpaired Student t-test).
|
 XML Download
XML Download