

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The increase in the global incidence of obesity over the past three decades runs parallel to an increase in the use of high-fructose corn syrup, which was first introduced in 1970 [1,2]. Obesity is a potent risk factor for hypertension, diabetes, and other metabolic diseases [3]. In animal models, several studies have indicated that high fructose intake leads to adverse metabolic and cardiovascular outcomes, including dyslipidemia, insulin resistance, hypertension, and weight gain [4,5]. In several studies, results have shown that high fructose intake induces hypertension in adult rats [6]. However, we are yet to understand the mechanisms by which high fructose intake induces hypertension.
The renin-angiotensin system (RAS) is a major regulator of blood pressure; electrolyte balance; and neuronal, renal, and cardiovascular control-related endocrine functions [7]. Angiotensin II (Ang II) is known to be produced systemically, and it plays a key role in the regulation of cardiovascular homeostasis [8]. Angiotensinogen (AGT) which is a substrate of RAS is released from the liver and is then cleaved in the circulatory system by renin. Renin is released from the juxtaglomerular apparatus of the kidney, to form angiotensin I (Ang I). Ang I is converted into Ang II by angiotensin-converting enzyme (ACE), which is largely expressed at high concentration on the surface of endothelial cells in the pulmonary system [8]. Ang II exerts its effects by stimulating specific receptors on the membranes of several organs. The effects of Ang II are mediated by two receptors, Ang II type-1 (AT1) and type-2 (AT2) receptor subtypes [9]. Activation of AT1 receptors leads to vasoconstriction, stimulation of catecholamine and antidiuretic hormone release, and production of thirst. This also promotes the growth of vascular and cardiac muscle [9]. In murine species, the AT1 receptor is subdivided into AT1A and AT1B subtypes [10]. The AT1A receptor is the most important of the subtypes in controlling blood pressure [11]. Systolic blood pressure is remarkably reduced in mice lacking AT1A receptors, while no significant difference in blood pressure between wild type mice and AT1B receptor knock out mice [12-14].
Ang II stimulates the secretion of the hormone, aldosterone, from the adrenal cortex [15,16]. Aldosterone lead to renal tubules to increase the reabsorption of sodium and water into the blood, while also causing the excretion of potassium to maintain electrolyte balance [17]. This increases the amount of extracellular fluid in the body, which also raises blood pressure [14,18]. However, there are no studies investigating whether a high-fructose diet can induce hypertension, RAS activation, and AT1R expression. We hypothesized that high fructose intake induces activation of RAS, resulting in hypertension and metabolic syndrome.
METHODS
Animals
This study was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals after approval by the Institutional Review Board of Kyungpook National University. Every effort was made to minimize both the number of animals used and their suffering. Thirteen-week-old male Sprague–Dawley (SD) rats were unilaterally nephrectomized under ketamine (150 mg/kg; Yuhan, Seoul, Korea) and xylazine (18 mg/kg; Bayer, Seoul, Korea) anesthesia, as previously described [19]. Eleven-week-old male SD rats were fed either a chow diet (control, n = 6) or a chow diet supplemented with 20% fructose (fructose, n = 6) in drinking water for two weeks [20-23]. Fructose was purchased from Millipore (Billerica, MA, USA). Rats were anesthetized with sodium pentobarbital (50 mg/kg intraperitoneally). Tissues were frozen in liquid nitrogen and stored at –80°C until further study. The number of approved animal study is KNU-2018-0145-1. We calculated the appropriate number of rats for this experiment by using the G*Power software.
Metabolic cage studies
Animals were housed individually in metabolic cages (Jeung Do Bio & Plant Co. LTD., Seoul, Korea) twice a week. We repeated the use of the metabolic cages four times. Food intake was calculated by subtracting the amount of food left over in each cage barrier for each rat from the measured amount of food provided on the previous day (g/kg/day). Average food intake values were presented as g/kg/day, and the body weight for each rat was determined twice a week (g). Water intake was calculated by subtracting the amount of water left over in each cage barrier for each rat from the measured amount of water provided on the previous day (ml/day). Average water intake values were presented as ml/day. Twice a week, rats were placed in metabolic cages for 24-h urine collection.
Glucose tolerance test (GTT)
Glucose tolerance tests were performed after fructose supplementation for 1 and 2 weeks. Rats were fasted 16 h before GTT experiments. Fasting glucose levels were determined using Accu-Chek Performa (Roche, Berlin, Germany). Glucose (50% solution, 2 g/kg) was injected intraperitoneally, and then, blood glucose levels were measured at 30, 60, and 120 min.
Blood pressure measurement
Blood pressure was measured in rats using the tail cuff method. Rats were preheated on a hotplate at 35°C for 10 min and then placed in plastic restrainers. A cuff with a pneumatic pulse sensor was attached to the tail. Blood pressure values were recorded on an NIBP Controller system (AD Instruments Pty Ltd., Castle Hill, Australia) with heating and were averaged from at least five consecutive readings obtained from each rat.
Quantitative reverse transcription polymerase chain reaction (RT-qPCR)
RT-qPCR was performed using an ABI PRISM 7000 Sequence Detection System (Applied Biosystems, Foster City, CA, USA), as previously described [19]. Amplification reactions were performed in a total volume of 20 μl, containing 10 μl of SYBR Green PCR Master Mix (Applied Biosystems), 4 μl of cDNA, and the indicated primer set (200 nM). All samples were amplified in triplicate in a 96-well plate, with the following cycling conditions: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 95°C for 15 s and 60°C for 1 min. Relative mRNA expression levels were determined by first calculating Δ cycle threshold (ΔCt) values by normalizing the average Ct value to its endogenous control (Gapdh) Ct value and then calculating 2–ΔΔCt. Statistical analyses were performed using one-way analysis of variance (ANOVA). The primer sets used for RT-qPCR are presented in Table 1.
Hormone measurements
Serum renin, Ang II, and aldosterone were analyzed individually by ELISA (E-EL-R0030, E-EL-R1430, E-EL-0070; Elabscience Biotechnology Inc., Houston, TX, USA).
Histological analysis
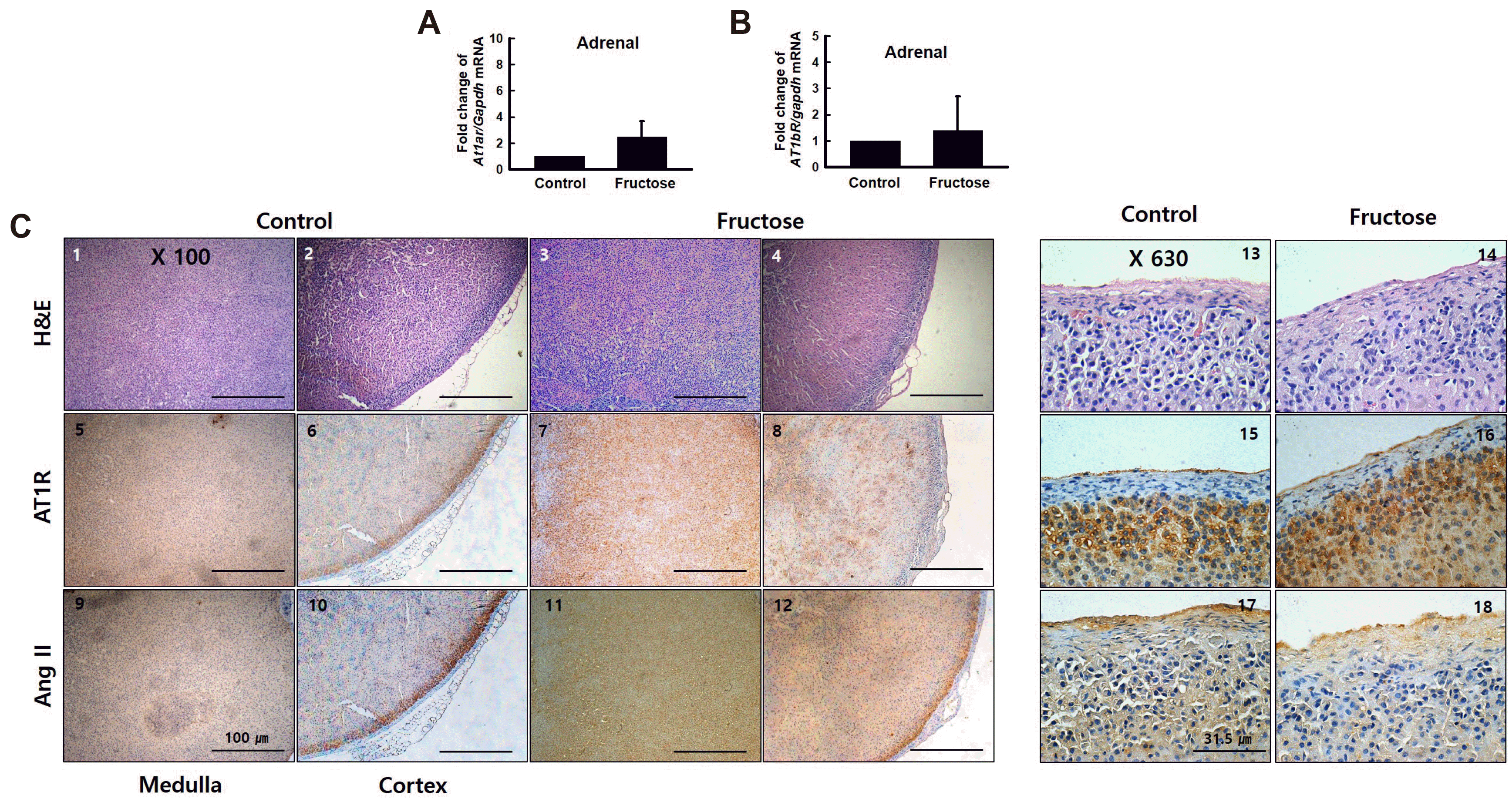
Kidney and adrenal gland tissues were fixed overnight in 4% formalin, dehydrated, and embedded in paraffin, using a conventional method. Paraffin-embedded samples were sectioned at a thickness of 3 μm. Sections were stained with hematoxylin and eosin (H&E) and trichrome. For immunohistochemistry, slides were incubated overnight with anti-AT1R or anti-Ang II antibodies at 4°C. The anti-renin antibody (PA5-21690) was obtained from Thermo Fisher (Waltham, MA, USA) and anti-AT1R (ab18801) and anti-Ang II (ab89892) antibodies were obtained from Abcam (Cambridge, UK). After staining, slides were examined using light microscopy.
Statistics
Results were expressed as mean ± standard error. Data were analyzed with the Kruskal–Wallis test or one-way ANOVA, followed by a post-hoc Tukey’s comparison test. Differences were considered significant at p < 0.05. A Student’s t-test was used to analyze differences between two groups. Statistical procedures were performed using SPSS software (release 19.0; IBM Co., Armonk, NY, USA).
RESULTS
High fructose intake increased body weight and water retention
We measured body weight in the two groups of rats. At the beginning of the diet, there were no significant differences in body weight between groups. The body weight of control rats increased normally. Intake of 20% fructose for two weeks significantly increased body weight. At the end of the experiment, there were significant differences in body weight between groups (Fig. 1A). Daily food intake (Fig. 1B) was not significantly different between the two groups. However, urine volume (Fig. 1D) decreased in the fructose group, and daily water intake (Fig. 1C) and water retention (Fig. 1E) significantly increased in the fructose group. These results suggested that high fructose intake increased body weight and water retention, regardless of food intake.
High fructose-induced hyperglycemia and hypertension
We measured glucose tolerance and blood pressure weekly. After two weeks of high fructose intake, systolic blood pressure was significantly elevated (Fig. 2A). However, there was no significant difference in fasting blood glucose levels between the two groups at week one or week two. Blood glucose levels after glucose loading were significantly different between the groups at week one and week two (Fig. 2B, D). High fructose intake resulted in significantly higher blood glucose levels and AUC values compared with the control diet (Fig. 2C, E). These findings suggested that high fructose intake induced metabolic syndrome.
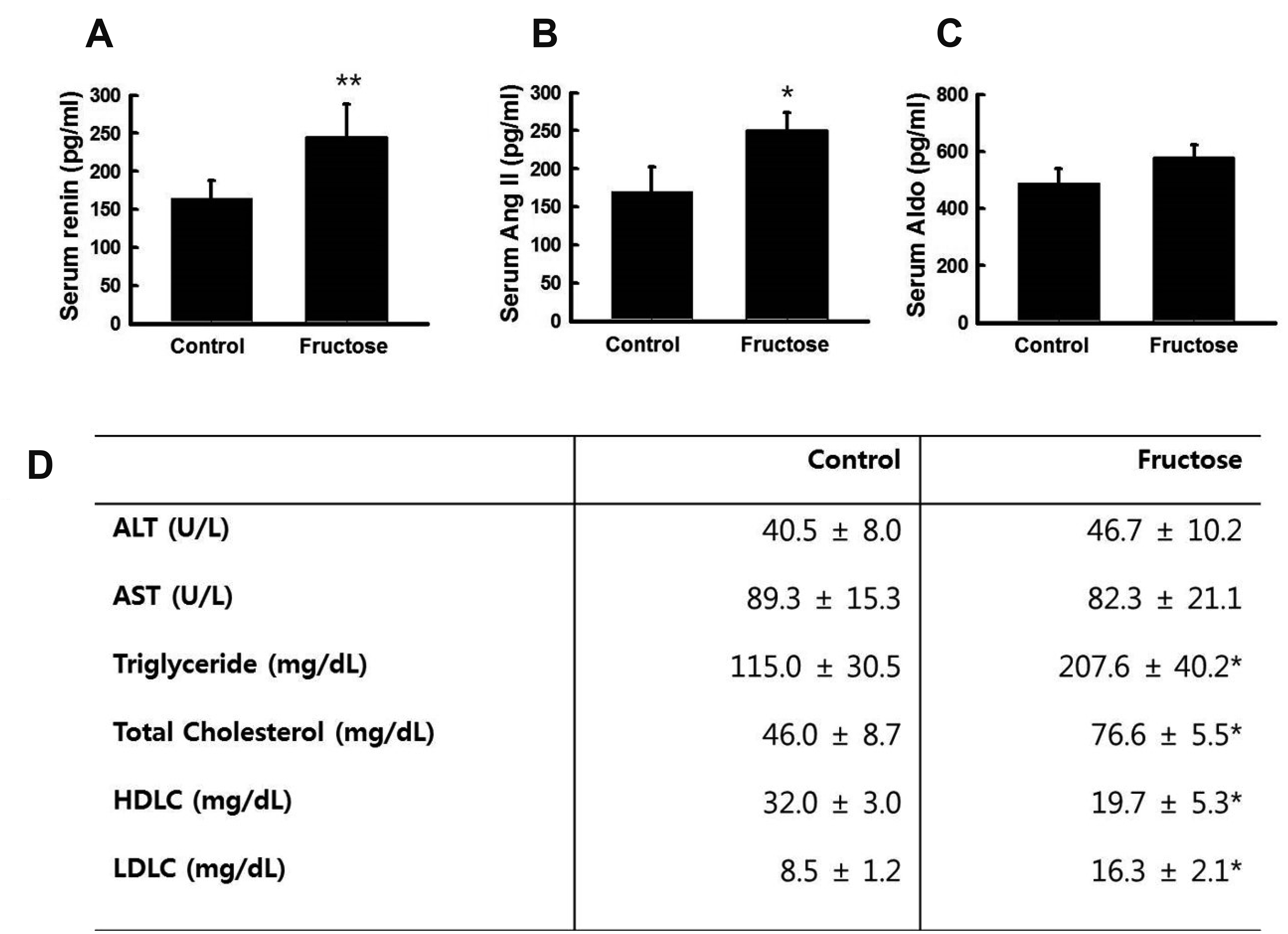
High fructose intake increases serum renin and Ang II levels and blood lipid concentration
To determine if a high-fructose diet had any effect on serum renin, Ang II, and aldosterone (Aldo) levels, we measured these parameters using an ELISA kit. Blood chemistry assays showed that serum levels of renin (Fig. 3A) and Ang II (Fig. 3B) were significantly elevated by fructose supplementation. However, serum Aldo levels were not affected by fructose intake (Fig. 3C). The fructose group had significantly higher blood triglyceride and cholesterol levels than the control group (Fig. 3D). These results suggested that high fructose intake increases renin and Ang II levels and blood lipid concentration.
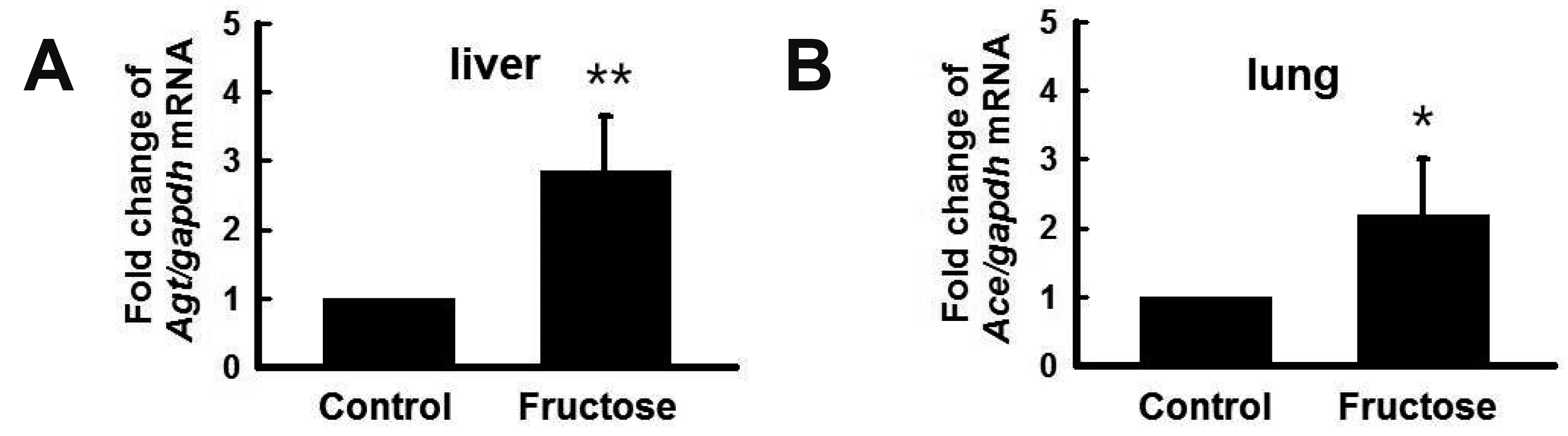
High fructose intake increased Agt and Ace expression
We analyzed the expression of the RAS genes, Agt and angiotensin-converting enzyme (Ace), by RT-qPCR. Fructose intake increased the expression of Agt in the liver (Fig. 4A) and Ace in the lungs (Fig. 4B). These findings indicated that high fructose increased RAS gene expression in the liver and lungs.
High fructose intake increased AT1R and Ang II protein levels in the kidneys
To determine whether fructose intake induced atrophy or hypertrophy in the kidneys and adrenal gland, we performed histological examinations. Fructose supplementation did not affect kidney or adrenal gland histology (Fig. 5A). We analyzed the expression of the RAS genes, renin, At1ar, At1br, and At2r by RT-qPCR. In the kidneys, fructose intake increased the expression of renin, At1ar, At1br, and At2r (Fig. 5B–E). However, the increase in At2r is thought to be a compensatory action to attenuate the increase in blood pressure induced by fructose. To analyze kidney morphology, we performed H&E staining (Fig. 5F; 1, 2) and immunohistochemical staining for renin (Fig. 5F; 3, 4), AT1R (Fig. 5F; 5, 6), and Ang II (Fig. 5F; 7, 8). Images from immunohistochemistry experiments indicated that renin, AT1R, and Ang II levels were elevated in the kidneys of rats in the fructose group. We also examined collagen deposition and fibrosis, which appear blue after trichrome staining. Fructose intake did not affect collagen deposition and fibrosis in the kidney (Fig. 5F; 9, 10). These results suggested that high fructose intake induced activation of RAS in the kidneys.
High fructose intake has no effect on the adrenal glands
We analyzed the expression of the RAS genes, At1ar and At1br, by RT-qPCR. The expression of At1ar and At1br in the adrenal glands did not increase with fructose intake (Fig. 6A, B). Images from immunohistochemistry experiments indicated that AT1R and Ang II protein levels were not elevated in the adrenal glands of rats in the fructose group (Fig. 5C). These results suggested that high fructose intake induced renin, AT1R, and Ang II activation in the kidneys but not in the adrenal glands.
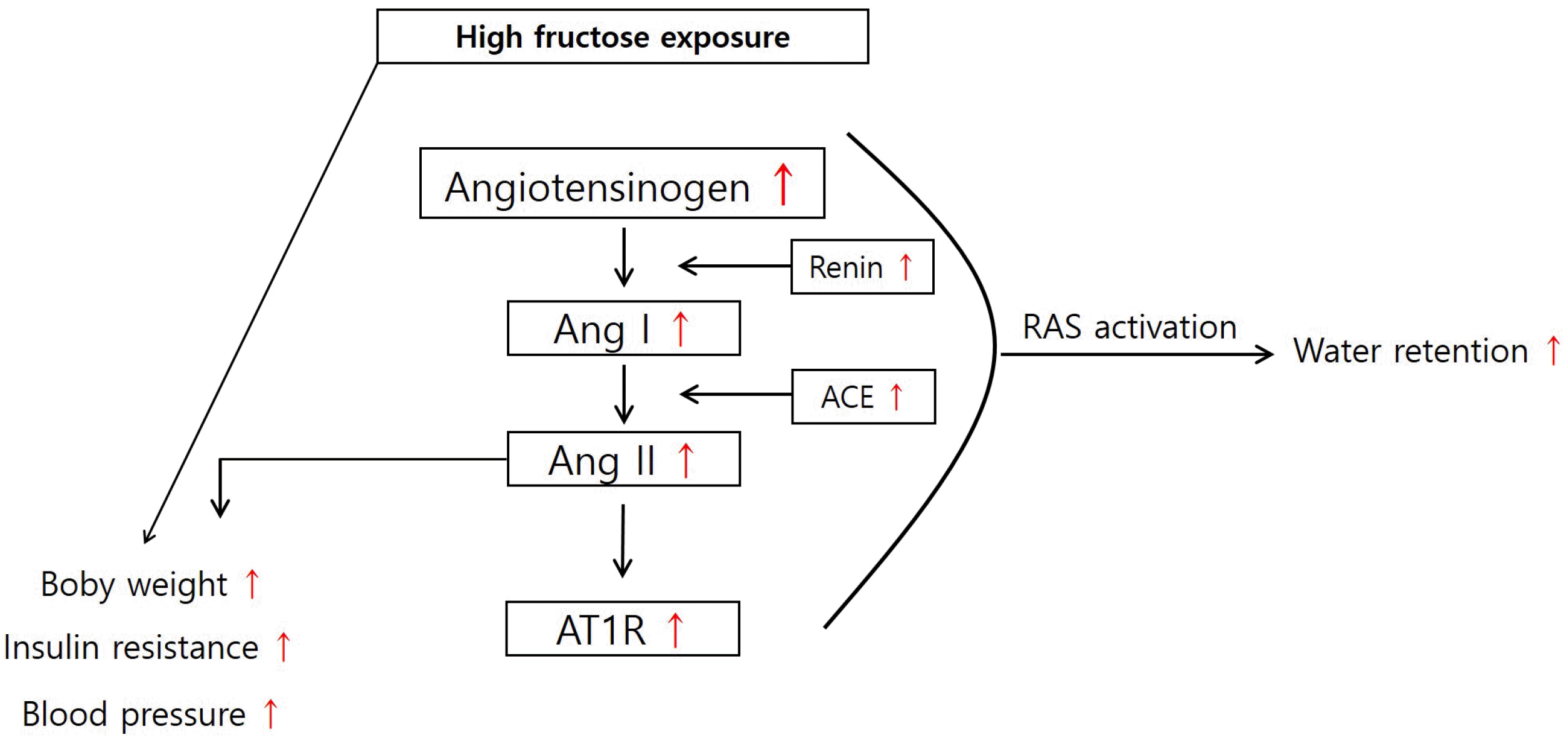
Schematic diagram
Eleven-week-old male SD rats were fed either a chow diet (control, n = 6) or a chow diet supplemented with 20% fructose (fructose, n = 6) in drinking water for two weeks (Fig. 7). High fructose intake induced activation of RAS, resulting in hypertension and metabolic syndrome.
DISCUSSION
In this report, we demonstrated that high fructose intake induced activation of RAS, resulting in hypertension and metabolic syndrome. We showed that high fructose intake increased body weight gain and water retention, regardless of food intake. High fructose intake induced hypertension and glucose intolerance after two weeks and activated serum renin and Ang II. Fructose is a simple monosaccharide found in many plants, but the two main sources of fructose in the western diet are sucrose and high-fructose corn syrup. Sucrose is a disaccharide with one molecule of glucose and one molecule of fructose, whereas high-fructose corn syrup is a mixture of glucose and fructose in varying ratios. Previous report showed that sucrose increased body weight and triglyceride [24]. Also, fructose may stimulate the production of advanced glycation end products (AGEs) that are toxic in diabetes [25].
Fructose intake also increased the expression of Agt in the liver and Ace in the lungs. In the kidneys, fructose intake increased the expression of renin, At1ar, At1br, and At2r. However, the expression of At1ar and At1br in the adrenal glands did not increase in the fructose group. In addition, fructose intake increased renin, AT1R, and Ang II protein levels in the kidneys, as detected by immunohistochemistry.
Consumption of sweetened beverages is associated with excessive caloric intake and an increased risk of diabetes and cardiovascular diseases due to an increase in weight [26]. Fructose plays an important role in the development of several metabolic disorders, and it has been implicated as the cause of a number of metabolic symptoms [27]. Several studies have demonstrated that high fructose intake is associated with increased development of metabolic syndrome components, such as hypertension, glucose intolerance, and weight gain [28,29]. In the current study, we showed that fructose intake induced weigh gain, glucose intolerance, and hypertension (Figs. 1 and 2).
Activation of the sympathetic nervous system results in an increase in renal renin release, leading to the generation of Ang I, which is then converted to Ang II, the major effector of RAS. This relationship between insulin resistance after a high fructose diet [30] and hypertension has been previously reported [24]. Based on this result, we analyzed blood chemistry and showed that serum levels of renin and Ang II, but not Aldo, were significantly elevated by fructose supplementation. Classically, aldosterone contributes to the regulation of blood pressure through its interaction with the mineralocorticoid receptor [31]. Aldosterone also exacerbates glucose intolerance induced by a high-fructose diet [32]. After six months of fructose intake, rats show significantly elevated serum aldosterone levels [33]. In our short-term study, serum aldosterone levels were not affected by two weeks of fructose intake. Fructose intake has been shown to increase the expression of RAS genes in the heart and aortae of male rats [34]. In the current study, high fructose intake resulted in an increase in the expression of the RAS genes, Agt in the liver (Fig. 4A), Ace in the lungs (Fig. 4B), renin in the kidneys (Fig. 5B), and AT1R in the kidneys (Fig. 5C, D), but not in the adrenal glands (Fig. 6). We also showed that high fructose intake resulted in an increase in the expression of At2r (Fig. 5E). There are several reports that At2r is upregulated in hypertension models, probably as a compensatory mechanism in response to increased renin-angiotensin activity [35,36]. At2r overexpression protects the heart from maladaptive remodeling and dysfunction of post-myocardial infarction [37]. AT2 receptor provides protection against AT1 receptor-mediated pressure and chronotropic effects [38]. The rise in uric acid in response to fructose may cause an afferent arteriolopathy resulting in glomerular hypertension [39]. Fructose is also filtered into the tubule where it is taken up in the S3 segment of the proximal tubule, leading to the local intracellular generation of uric acid with oxidative stress and local inflammation [40]. In our study, the kidney is directly involved in the reabsorption of water. For two weeks, the amount of water consumed increased whereas the volume of urine decreased due to the intake of fructose (Fig. 1). Increasing plasma osmolality is a cause of volume overloading and activation of RAS. Therefore, we think RAS was activated before adrenal glands by hypertrophy of the kidney (Fig. 5).
Another group reported that fructose via uric acid stimulates renal expression of pro-renin-receptor (PRR)/soluble PRR that stimulate sodium/hydrogen exchanger 3 and Na/K/2Cl cotransporter expression and intrarenal renin–angiotensin system to induce salt-sensitive hypertension [41]. Both full-length PRR and soluble PRR are able to bind to renin and the inactive prorenin, thus enhancing the catalytic activity of renin, promoting the nonproteolytic activation of prorenin, and increasing the catalytic efficiency of local Ang II formation. Activation of the sympathetic nervous system increases renin release through the activation of β2-adrenergic receptors, which leads to the sequential generation of Ang I and Ang II, the latter being a major effector of RAS.
The recent publication reported that fructose contributes to hypertension by stimulating NKCC2-dependent NaCl reabsorption in thick ascending limb [42]. Furthermore, another group reported that Fructose stimulates Na/H exchange activity and sensitizes the proximal tubule to Ang II [43].
Aldosterone is released from the adrenal cortex. We confirmed that fructose had no effect on the activation of AT1R or Ang II in the adrenal glands. In accordance with this result, serum aldosterone levels were also not affected by two weeks of fructose intake. Taken together, these results indicated that fructose intake induced hypertension, RAS activation, and elevated expression of AT1R in the kidneys. The results of the present study are summarized in Fig. 7.
In summary, this study demonstrated that a high-fructose diet induced metabolic syndrome symptoms, such as body weight gain, insulin resistance, and high blood pressure. A high-fructose diet also induced AT1R and Ang II activation in the kidneys but not in the adrenal glands. Specifically, our study suggests that the development of hypertension due to the consumption of fructose-containing beverages may be associated with RAS. Therefore, AT1R inhibition may be a potential therapeutic target for preventing fructose-induced hypertension.

 XML Download
XML Download