

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
The vascular endothelium is defined as a single layer of endothelial cells (ECs) that line the lumen of blood vessels and are mechanically and metabolically dynamic organs. The endothelium, which consists of 1 – 6 × 1013 individual ECs, is the largest organ in the body and exceeds 1,000 m2 of estimated surface area [1-3]. This important organ is involved in a variety of physiological and pathological functions including blood supply, nutrient delivery, immune cell adhesion, vasopermeability, angiogenesis, thrombogenesis, and vascular tone [4-7].
Endothelium-dependent vasodilation is largely determined by alterations in endothelial intracellular Ca2+ concentrations in response to mechanical stimuli (e.g., shear stress, membrane stretch) or endogenous agonists (e.g., bradykinin, ATP, or reactive oxygen species [ROS]). Increased intracellular Ca2+ levels produces nitric oxide (NO) and prostacyclin (PGI2) that are traditionally considered as endothelium-derived relaxing factors (EDRFs) [8,9]. In addition, it has been well documented that changes in global or localized EC Ca2+ signaling stimulate Ca2+-sensitive K+ channels and elicit the membrane hyperpolarization of ECs and vascular smooth muscle cells (VSMCs) in a sequential manner [10]. This discovery contributes to the development of a novel concept, endothelium-dependent hyperpolarization (EDH), which is a primary mechanism of vasodilation in small resistance arteries.
It is no exaggeration to state that the elucidation of EC Ca2+ signaling has been accomplished by the progression of sophisticated imaging techniques and fast/high-affinity fluorescent Ca2+ indicators, including a genetically modified mouse specifically expressing a Ca2+ indicator (e.g., GCaMP2) in ECs, high-quality (i.e., high speed and resolution) confocal microscopy, and total internal reflection fluorescent (TIRF) microscopy [11]. These technological advances have led investigators to explore complicated Ca2+ dynamics in ECs including Ca2+ release from the endoplasmic reticulum (ER; i.e., propagated Ca2+ wave [12], Ca2+ pulsars [13], Ca2+ wavelets [14]) and Ca2+ entry from the extracellular space (i.e., Ca2+ sparklets [15]) that result in vasodilation. However, the molecular mechanisms underlying the Ca2+ influx in ECs and their regulations have been poorly defined. In light of this, the identification of transient receptor potential (TRP) channels has provided new insights into Ca2+ mobilization in ECs that is required for EDH(F) and vasodilation. Thus, this review focuses on describing the contribution of TRP channels to fundamental Ca2+ signals (i.e., serve as a crucial means of altering intracellular Ca2+ levels) in the ECs of resistance arteries.
Go to : 
Ca2+ SIGNALING AND EDH
The discovery of NO and PGI2 generated from ECs has provided insight into the novel paradigm that the endothelium is an organ that does not merely cover the inner wall of blood vessels; it also controls the vascular tone and blood flow [10]. In addition, succeeding investigations have shown that EC-dependent VSMC hyperpolarization (caused by releasing factors from the ECs: endothelium-dependent hyperpolarizing factors [EDHFs]) in response to muscarinic receptor activation elicits vasodilation by inhibiting voltage-dependent Ca2+ channels (VDCCs) in VSMCs [16,17]. Chen and colleagues [17] sought to directly assess the effects of acetylcholine (ACh) on the membrane potential of VSMCs in the aorta and main pulmonary artery of rats. ACh-induced VSMC hyperpolarization was still detected even in the presence of inhibitors of NO or guanylyl cyclase, suggesting that EDHFs are distinct from EDRFs. Importantly, that study demonstrated that K+ efflux is a key component of EDHFs [17]. Since then, the previous findings on EDHFs have evolved into a new concept of EDH; additionally, such studies have seminally identified intermediate/small conductance Ca2+-sensitive K+ channels and microdomain structures (e.g., myoendothelial projections [MEPs] and myoendothelial gap junctions [MEGJs]) that allow the movement of hyperpolarizing currents from ECs to VSMCs [18].
In the Garland and McPherson study [19], although VSMC hyperpolarization in response to NO was significantly attenuated by a K+ channel blocker (i.e., glibenclamide), ACh-stimulated hyperpolarization was not affected by the same blocker. The hyperpolarization and vasorelaxation induced by ACh were not sensitive to NO synthase (NOS) or cyclooxygenase (COX) inhibitors (i.e., L-NNA or indomethacin, respectively) in rat small mesenteric arteries [19]. These results suggest that ACh-mediated VSMC hyperpolarization leads to vasorelaxation that occurs independently of NO or prostaglandins. Further, subsequent works have demonstrated that ACh-induced hyperpolarization and vasorelaxation are associated with the activation of Ca2+-sensitive IKCa and SKCa channels distributed in ECs [20-22]. This finding implies that muscarinic receptor activation evokes inositol trisphosphate (IP3)-mediated Ca2+ release from the ER and activates IKCa/SKCa channels. Another investigation in rat hepatic arteries showed that K+ efflux from ECs through IKCa/SKCa channels acts as an EDHF by activating inwardly rectifying K+ (Kir) channels and Na+-K+ ATPase in adjacent VSMCs [23]. However, since blockers of Kir channels and Na+-K+ ATPase (Ba2+ and ouabain, respectively) did not entirely abolish vasorelaxation in rat small mesenteric arteries, EC hyperpolarization might be directly transmitted to the VSMCs through MEGJs [23].
It is generally accepted that the vasorelaxation or dilation of small resistance arteries primarily depends on EDH rather than on EDRFs (e.g., NO, PGI2) or EDHFs [18]. MEPs are defined as extensions of ECs that project through the fenestration of the internal elastic lamina (IEL) to adjacent VSMCs. The MEGJs located at the tip of the MEP are intercellular pores that allow the spread of second messengers (e.g., IP3) or charged ions for heterocellular (EC-VSMC) communication [24]. IKCa and SKCa channels that are indispensable for EDH-mediated vasodilation in resistance arteries are activated by the binding of the Ca2+-calmodulin complex to the C-terminus of those channels [25]. K+ loss via the Ca2+-sensitive K+ channels elicits hyperpolarization that is conducted to VSMCs through MEGJs, which leads to vasodilation by inhibiting VDCCs (Fig. 1). Thus, it is strongly suggested that EC Ca2+ signaling, in particular at MEPs, plays a critical role in EDH-dependent vasodilation. Indeed, since it was first discovered that an alteration in intracellular Ca2+ concentrations is a major determinant of vascular reactivity [26], considerable strides have been made in understanding the role of EC Ca2+ signaling in EC-dependent vasodilation in resistance arteries. According to previous studies, cation-permeable TRP ion channels that allow for Ca2+ entry into ECs have been shown to induce EDH-mediated vasodilation in small arteries [13-15,27,28]. Thus, EC TRP channel-dependent Ca2+ signaling will be discussed in this review.
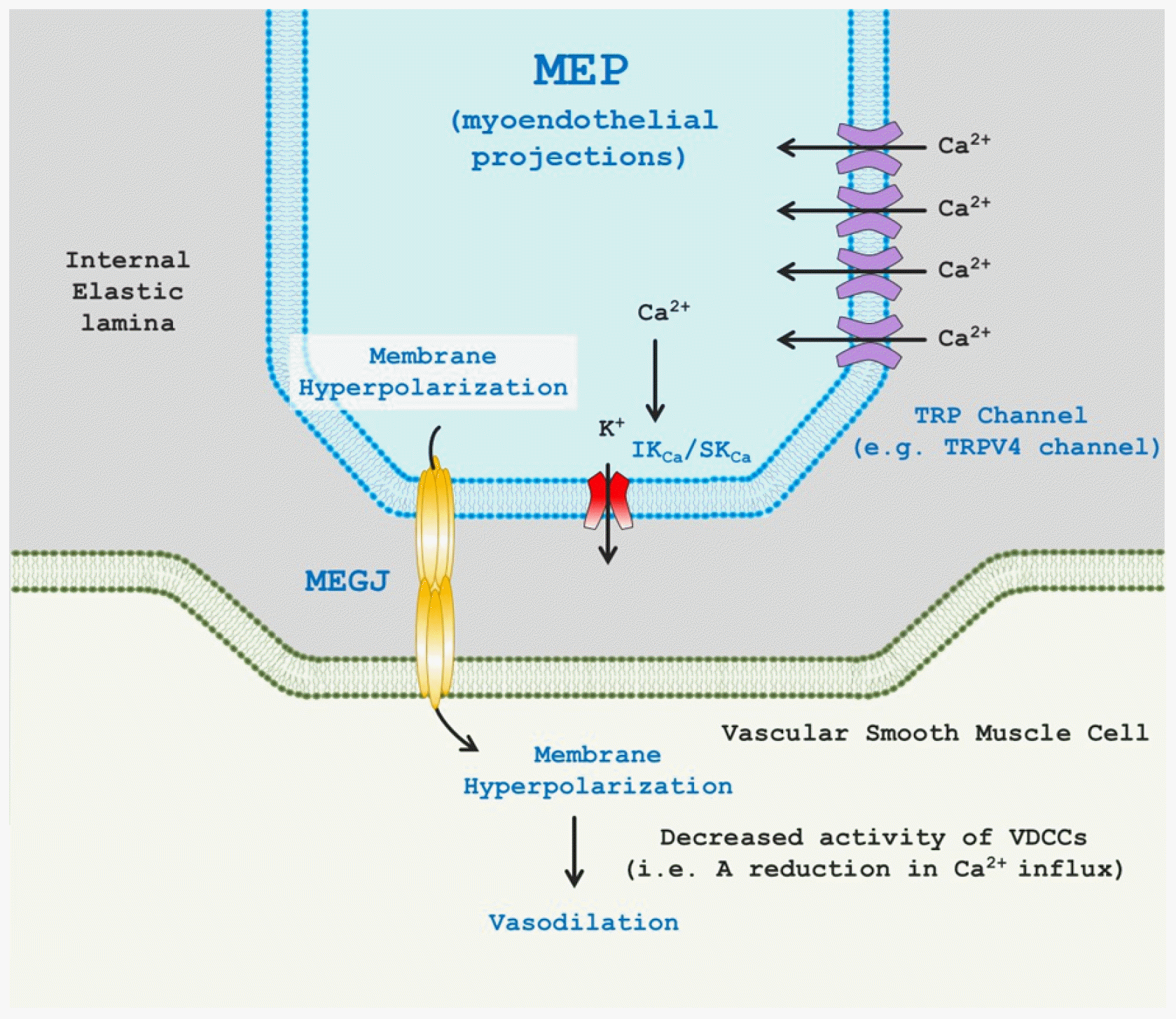 | Fig. 1Transient receptor potential (TRP) channel-mediated endothelium-dependent hyperpolarization (EDH) and vasodilation.TRP channels (e.g., representatively TRPV4 channels) are placed myoendothelial projections (MEPs) that are extensions of endothelial cells (ECs) through internal elastic lamina to contact adjacent vascular smooth muscle cells (VSMCs). Localized Ca2+ entry through endothelial TRP channels stimulate intermediate/small conductance Ca2+-sensitive potassium (IKCa/SKCa) channels and in turn results in EDH via K+ ion efflux. EDH is transmitted to adjacent VSMCs through myoendothelial gap junctions (MEGJs) comprising of two hemi-channels, which initiates VSMC hyperpolarization. The altered membrane potential of VSMCs suppresses voltage-dependent Ca2+ channels (VDCCs) and evokes vasodilation.
|
Go to :
Ca2+ INFLUX THROUGH TRP CHANNELS AND SUBSEQUENT VASODILATION
The TRP superfamily is encoded by 28 genes and is classified according to DNA and protein sequence homology. Consequently, TRP channels are categorized into six subfamilies: TRPA (ankyrin), TRPC (canonical), TRPM (melastatin), TRPML (mucoliptin), TRPP (polycystin), and TRPV (vanilloid) channels. TRP channels are generally comprised of seven hydrophobic domains. Six (S1–S6) of the domains span the plasma membrane, and the seventh domain intracellularly exists with the COOH- and NH2-termini of TRP channels [29-31]. The ion-conducting pore region is located between the fifth and sixth domains (S5 and S6 domains). The TRP box, a conserved 25-amino acid element, is formed immediately after the S6 domain in the subfamilies of TRPC, TRPM, and TRPV channels [32]. Although the TRP domain has been incompletely examined, it has been elucidated to date that the conserved box serves as a platform for a phosphatidylinositol 4,5-bisphosphate (PIP2) binding site or ion channel heteromeric assembly [33,34]. The intracellular C- and N-termini of TRP channels exist for multiple regulatory elements, interaction sites, and enzymatic domains [32].
TRP channels are postulated to participate in diverse physiological processes (e.g., mechanotransduction, cellular excitability, protein trafficking, and vascular contractility) by regulating cation influx across the cell membrane [32]. TRP channels physiologically control the membrane potential and regulate Ca2+ signaling in ECs and VSMCs. Among TRP channel subfamilies, Na+-permeable TRPM4 channels (equally permeant to K+, but not Ca2+-permeable) are activated by globally or locally increased intracellular Ca2+ levels in VSMCs [35]. The activated channels primarily elicit inward currents of Na+ in cerebral VSMCs, induce membrane depolarization, and subsequently activate VDCCs [36]. In addition to membrane potential regulation, Ca2+ signaling that is required for vasoconstriction or dilation is controlled by Ca2+-permeable TRP channels in a direct or indirect (i.e., metabotropic) manner. For example, Ca2+ influx via the TRPV4 and TRPA1 channels in ECs that are directly stimulated by shear stress, endogenous lipid arachidonic acid, or exogenous agonists (e.g., GSK1016790A or AITC) triggers the vasodilation of cerebral or mesenteric arteries [15,37-39].
Localized Ca2+ signaling through endothelial non-selective cation TRP channels primarily plays a vital role in the generation of EDH via the downstream activation of IKCa/SKCa channels and vasodilation in small-sized resistance arteries. It is well documented that vascular peripheral resistance is a major determinant of arterial pressure and blood flow to organs and tissues. Furthermore, vascular resistance is regulated by EDH and subsequent endothelium-dependent vasodilation in the microcirculation. Indeed, impaired EDH and endothelium-dependent vasodilation in microvascular beds is found to cause cardiovascular diseases including hypertension. Thus, this review mainly focused on TRP channel-mediated EDH in resistance arteries as they are physiologically significant in the regulation of vascular resistance, intravascular pressure, and blood flow.
TRPC1
TRPC1 channels are non-selectively permeable to Ca2+ (a unitary conductance of ~5pS) [40]. Although TRPC1 channel is known to be expressed in the endothelium, it is debatable that TRPC1 channels show homomeric channel in intact endothelial cells. In contrast, it is definitely elucidated that TRPC1 channels form heteromeric channels with TRPC3 [41], TRPC4 [40], TRPC5 [40], and TPRV4 [42,43] channels. With regard to the role of TRPC1 channels in endothelial intracellular Ca2+ signaling, the activation of Ca2+-sensing receptor (CaSR) stimulates Ca2+ influx through TRPC1 channels and leads to NO production in the endothelium [44]. Furthermore, once CaSR is activated by the elevation of extracellular Ca2+ concentration, TRPC1 channels that are co-localized with TRPV4 channels showed vasorelaxation, which was significantly abrogated in the presence of TRPC1 and TRPV4 inhibitors [42]. Therefore, it is suggested that the heteromeric TRPC1/TRPV4 channels stimulated by CaSR activation induce NO-mediated vasorelaxation.
TRPC3
TRPC3 channels are expressed in arteriolar myocytes and contribute to membrane depolarization-induced vasoconstriction (i.e., permeability: Ca2+ = Na+ > K+) [45-48]. Meanwhile, growing evidence has shown that TRPC3 cation channels are expressed in ECs and are responsible for EC hyperpolarization-mediated vasodilation. The vasoreactivity of rat small mesenteric arteries was considerably blunted in response to flow or bradykinin in the treatment of TRPC3 antisense oligonucleotide [49]. Subsequent pharmacological studies with a specific inhibitor of TRPC3 channels (i.e., Pyr3) have consistently demonstrated that the ACh-induced vasodilation of human internal mammary arteries or rat mesenteric arteries is markedly reduced with Pyr3 pretreatment [50,51], implying that EC GPCR activation by shear stress or exogenous agonists is coupled to TRPC3-induced downstream signaling for vasodilation. Senadheera and colleagues used ultra-structural immunohistochemistry to show that TRPC3 channels are spatially restricted at MEPs (i.e., five-fold higher expression compared to non-MEPs) in resistance mesenteric arteries [51]. This investigation also revealed that TRPC3 inhibition with Pyr3 causes a significant reduction in EC hyperpolarization in response to ACh [51]. More recently, studies have shown that EC TRPC3 channels in cerebral arteries are translocated to the plasma membrane following purinergic receptor activation with ATP and are then involved in EDH-mediated vasodilation [52]. Collectively, it is speculated that second messengers (e.g., DAG) produced by GPCR activation stimulate Ca2+ influx via TRPC3 channels and in turn lead to EDH-mediated vasodilation in resistance small arteries.
TRPC4
TRPC4 channels preferably allow Ca2+ influx in the endothelium through store-operated Ca2+ entry and are stimulated by GPCR downstream signaling [32,53]. Freichel and colleagues [54] demonstrated that endothelium-dependent vasodilation occurs through the activation of TRPC4 channels in mouse aorta. The specific molecular mechanism underlying the vasodilation is attributed to endothelial NO synthase (eNOS) activation and NO production followed by the activation of muscarinic acetylcholine receptors [54,55]. Even though subsequent studies failed to show that endothelial store-operated Ca2+ entry is mediated by TRPC4 channels [53,56], it is noteworthy to understand that TRPC4 channels could be involved in endothelium-dependent vasodilation in conduit arteries, but not small-sized resistance arteries.
TRPC5
TRPC5 channels show a unitary conductance of 40–60 pS and are more permeable to Ca2+ than Na+ (PCa:PNa = 1.8–9.5) [57,58]. Although the role of TRPC5 channels in endothelial function has not been clearly identified to date, previous investigations found that TRPC5 channels are distributed in the endothelium from mouse aorta, human coronary, and cerebral arteries [59-61]. Mori and colleagues revealed that Ca2+ influx through endothelial TRPC5 channel is regulated by post-translational modifications. Specifically, S-nitrosylation, the covalent attachment of an NO to cysteine groups, enhances the activity of TRPC5 channels and Ca2+ influx in cultured ECs [62]. However, the alteration of vasodilatory response to ACh was not monitored in mouse aortic rings originated from TRPC5 knockout models [60]. This suggests that NO-mediated vasodilation in the presence of ACh was not augmented by TRPC5 channels, which is inconsistent with previous studies [62]. Thus, it is necessary in future studies to explore if Ca2+ entry via TRPC5 channels elicits vasodilation and via what supporting mechanisms.
TRPM2
TRPM2 channels that have a similar permeability of Ca2+ and Na+ ions display the unitary conductance of 58–76 pS [63]. The subtype of TRPM2 channels is gated by several activators including ADP-ribose, arachidonic acid, hydrogen peroxide [64-66]. The Ca2+ influx of TRPM2 channels is also clarified to induce endothelium-dependent vasodilation. It was found in rat cremaster resistance arteries that exogenous hydrogen peroxide application leads to Ca2+ entry and subsequent vasodilation [67]. However, those Ca2+ signaling and vascular reactivity were markedly diminished by the treatments of TRPM2 antibody or IKCa/SKCa channel inhibitors. These results indicate that hydrogen peroxide-mediated Ca2+ entry through TRPM2 channels evokes endothelial vasodilation in the manner dependent of EDH [67].
TRPV1
TRPV1 nonselective cation channels are expressed in arterial ECs [68,69] and are favorably permeable to Ca2+ compared with Na+ (PCa:PNa ≈ 9.6) [32]. Capsaicin, endocannabinoids, and noxious heat activate TRPV1 channels [70,71]. Anandamide-evoked TRPV1 activation increased NO in the endothelium of rat mesenteric arteries [72]. A synthetic cannabinoid compound (VSN16) induced vasorelaxation of the mesenteric arteries, which was markedly attenuated by capsazepine (a TRPV1 antagonist), L-NNA (an NOS inhibitor), charybdotoxin (an IKCa/BKCa channel inhibitor), or apamin (a SKCa blocker) [73]. In gracilis skeletal muscle arterioles, the capsaicin-mediated stimulation of TRPV1 channels produced vasodilation that was substantially attenuated by L-NAME (an NOS inhibitor), which suggests that EC TRPV1-mediated vasodilation occurs in an NO-dependent manner [74]. Interestingly, NO-mediated vasorelaxation/dilation following TRPV1 activation differs from vasodilatory mechanisms of other TRP subfamilies (i.e., TRPV4, TRPA1) that display EDH-dependent vasodilation. In accordance with previous studies showing a relationship between EC TRPV1 activation and NO production, capsaicin treatment increased intracellular EC Ca2+ levels and elicited the phosphorylation of eNOS in cultured mouse aortic ECs [75]. Additionally, the chronic administration of capsaicin profoundly increased eNOS phosphorylation at Ser1177, NO generation, and protein kinase A (PKA) activation in mouse mesenteric arteries; these changes were absent in TRPV1 knockout mice [75]. As expected, the chronic activation of TRPV1 channels was coupled to the enhancement of EC-dependent vasorelaxation in freshly isolated mesenteric arteries [75]. Surprisingly, this study showed convincing clinical evidence that chronic capsaicin application significantly reduces blood pressure in spontaneously hypertensive rats. This finding implies that TRPV1 channels participate in the regulation of vascular tone and arterial pressure. The following study in bovine aortic ECs found that capsaicin-induced Ca2+ entry via TRPV1 channels stimulates Akt-calmodulin-dependent protein kinase II (CaMKII)-eNOS pathway; in turn, phosphorylated eNOS physically interacts with TRPV1 channels to produce NO [76]. However, despite consistent findings, a recent study elucidated that large renal arteries in mice do not respond to a relatively higher concentration of capsaicin (i.e., a 100-fold higher level of EC50 is required for significant vasodilation), whereas mesenteric small arteries showed a marked vasodilation in response to 25 nM of capsaicin [77]. Thus, the magnitude of TRPV1-dependent vasoreactivity is specific to vascular beds.
TRPV3
TRPV3 channels display a large unitary conductance (≈ 150–200 pS) [78]. Dietary monoterpenoid compounds including carvacrol, eugenol, and thymol stimulate TRPV3 channels in the oral/nasal epithelium [79]. With regard to the roles of TRPV3 in EC-dependent vasodilation, Earley and co-workers revealed for the first time that carvacrol that is typically found in the Mediterranean diet, induces vasodilation in rat cerebral arteries [80]. Increased endothelial Ca2+ concentrations and TRPV3-like currents have been found in the presence of carvacrol [80]. Moreover, TRPV3-dependent cerebral vasodilation was found to result from VSMC hyperpolarization that was inhibited by TRAM-34, apamin, or BaCl2 (an inhibitor of inwardly rectifying K+ (Kir) channels), but not by L-NNA or indomethacin. This suggests that TRPV3 activation-induced K+ efflux through IKCa or SKCa leads to the Kir-mediated hyperpolarization of VSMCs and cerebral vasodilation [80]. Recently, it was demonstrated that EC TRPV3 Ca2+ sparklets (i.e., localized Ca2+ influx through EC TRPV3 channels) are directly and optically detected with carvacrol in the ECs of rat cerebral parenchymal arterioles [27]. The unitary Ca2+ events in ECs were significantly attenuated by a selective TRPV3 antagonist, isopentenyl pyrophosphate (IPP). This study also noted that TRPV3 sparklet-mediated vasodilation is not sensitive to NOS or COX inhibitors. By contrast, the inhibition of IKCa or SKCa channels markedly reduced the dietary compound-induced vasodilation. Importantly, the authors also suggested that the amplitude of TRPV3 channels (∆F/F0 = 0.20; peak fluorescence [F] is normalized to baseline fluorescence [F0]) is greater than that of TRPA1 (∆F/F0 = 0.13) and TRPV4 (∆F/F0 = 0.06) channels. TRPV3 Ca2+ sparklets were implicated to play a key role in EC Ca2+ signaling in cerebral resistance arterioles [27]. However, to the best of our knowledge, there have been few observations on the influence of TRPV3 channels on vasomotor reactivity. Thus, further observations are required for a better understanding of the contribution of EC TRPV3 for the modulation of vascular function.
TRPV4
TRPV4 channels were shown to have a specific permeability to Ca2+, Na+, and Mg2+ (PMg:PNa ≈ 2–3:1; PCa:PNa ≈ 6–10:1) [68,81,82]. TRPV4 channels are stimulated by a variety of endogenous activators or synthetic agonists including 4α-phorbol 12,13-didecanoate (4α-PDD; 200 nM of EC50; [83]), GSK1016790A (18 nM of EC50; [84]), RN-1747 (4 μM of EC50; [85]), or arachidonic acid metabolites (e.g., 5,6-EET; 130 nM of EC50; [86]). When Ca2+-permeable TRPV4 channels were pharmacologically activated, increased whole-cell currents or Ca2+ influx were observed in isolated ECs [87-90]. Previous investigations demonstrated that in small arteries, EC TRPV4-mediated alterations in Ca2+ levels following the activation of muscarinic or purinergic receptors in ECs contribute to marked vasodilation. ACh fails to dilate mesenteric arteries with a genetic deletion of TRPV4 channels [90]. In this study, ACh-mediated Ca2+ influx and NO production were also significantly blunted in TRPV4-/- mice. Similarly, a potent TRPV4 antagonist (RN-1734) pretreatment considerably disrupted the ACh-evoked vasodilation of radial uterine arteries [91].
Sullivan et al. [92] and Sonkusare et al. [15] marked the beginning of a new area of endothelial Ca2+ influx through TRPV4 channels, called TRPV4 Ca2+ sparklets, by using improved and sophisticated optical imaging techniques including high-speed/resolution confocal or TIRF microscopy. The Sonkusare and Nelson group sought to delineate the properties of elementary TRPV4 Ca2+ sparklets spatially identified at MEPs. TRPV4 channels were revealed to exhibit quantal events of Ca2+ entry, and the quantal amplitudes were shown to be dependent on the electrochemical gradient. Surprisingly, three to eight TRPV4 sparklet sites per EC was considered firmly sufficient for maximal vasodilation [15,92]. Extensive monitoring also showed that Ca2+ signaling through EC TRPV4 channels is explicitly coupled to IKCa and SKCa activation-mediated hyperpolarization at MEPs and vasodilation [15]. Further, K+ efflux via IKCa and SKCa channels enhanced vasodilation by stimulating Kir channels in ECs and VSMCs and by amplifying the hyperpolarization of both cells [23,93]. Next, one of the novel characteristics of TRPV4 sparklets is channel cooperative behavior [94]. Ca2+-permeable TRPV4 channels are interestingly designed with a four-channel (tetramer) metastructure. The clustered TRPV4 channels locally distributed at MEPs are cooperatively gated, which is determined by protein kinase C-anchoring protein 150 (AKAP150) and intracellular Ca2+ [94]. It is conceivable that opening only one single TRPV4 channel may induce a minimal Ca2+ influx and activate a small number of IKCa and/or SKCa channels at the microdomains. However, increased cooperativity of the clustered TRPV4 channels (i.e., quantal events) gives rise to augmented Ca2+ amplitudes and enhanced hyperpolarization [94]. Angiotensin II-induced hypertension was shown to impair AKAP150-dependent cooperative activity of TRPV4 channels by disrupting the scaffolding protein expression at MEPs [94]. It is speculated that EC TRPV4 and AKAP150 play a critical role in preventing pathophysiological circumstances in the cardiovascular system.
With respect to the roles of TRPV channels in mechanotransduction, since ECs directly interact with shear stress induced by blood flow velocity and blood viscosity due to their unique anatomical position, considerable attention was given to flow (shear stress)-mediated EC TRPV4 activation and subsequent vasodilation in small resistance arteries. Further, because most previous studies of TRPV4 Ca2+ imaging were implemented in the presence of chemical agonist compounds (e.g., 4α-PDD, GSK1016790A), the impact of physiological modulators including shear stress or intravascular pressure on EC TRPV4 activation and downstream signaling for vasodilation requires further exploration. TRPV4 channels activated by shear stress resulted in increased Ca2+ entry in the ECs of mesenteric arteries and vasodilation that was significantly suppressed in TRPV4-deficient mice [38]. Pharmacological inhibition to NOS entirely suppressed shear stress (10 dyn cm-2)-mediated vasodilation in mesenteric arteries [38], which was consistently shown in gracilis muscle arterioles [88]. These studies suggest that the mechanoactivation of TRPV4 channels exerts vasodilation through NO production; however, it is not completely clear as to how the channels are directly or indirectly activated by shear stress or metabolites (e.g., epoxyeicosatrienoic acids [EETs]) produced by shear stress. Conversely, the perfusion of 4α-PDD and 11,12-EET still evoked vasodilation in mesenteric arteries even in the presence of NOS and COX inhibitors [72,95]. It is implied that different physiological or pharmacological forms of activation of TRPV4 channels may result in disparate downstream signaling for vasodilation [88], presumably due to the unique polymodal nature of TRPV4 channels. Further, it was delineated in human coronary arteries that flow-dependent activation of TRPV4 channels generates mitochondrial ROS (i.e., hydrogen peroxide and superoxide) that play an important role in vasodilation in human coronary arteries [96].
As mentioned above, although VSMCs are constantly exposed to intraluminal pressure, biological processes in ECs are mechanically affected by shear stress. Nonetheless, EC TRPV4 channel activity has been demonstrated to be modulated by low intravascular pressure (i.e., below 50 mmHg) in rat cremaster or mesenteric arterioles [97]. Specifically, the frequency of EC Ca2+ events via EC TRPV4 channels was further augmented at a low intraluminal pressure (two- to three-fold higher than those at 60–80 mmHg of arterial intraluminal pressure). Both pharmacological (i.e., selective TRPV4 agonists) and physiological (i.e., intraluminal pressure) TRPV4 activations were markedly diminished by RN1734 or HC067047 [97]. Although they revealed that TRPV4, IKCa, and SKCa channels are clustered at perforations in the IEL, the inhibition of IKCa with TRAM-34 (but not the inhibition of SKCa with apamin) considerably enhanced the basal myogenic tone in cremaster arterioles. Additionally, the blockade of PLC and the IP3 receptor with U-73122 and xestospongin fully abrogated the low pressure-mediated increase in Ca2+ sparklet events. Thus, Ca2+ influx via EC TRPV4 channels in response to low intraluminal pressure stimulates IKCa channels through PLC and IP3R signaling, which in turn leads to EC/VSMC hyperpolarization-dependent vasodilation [97].
Arachidonic acid and its metabolites are potential physiological modulators for the activation of TRPV4 channels in ECs. Arachidonic acid is converted to EETs such as 5,6-EET, 8,9-EET, 11,12-EET, and 14,15-EET through cytochrome P450 (CYP) epoxygenase [86]. It has been well known that EETs have anti-hypertensive/inflammatory effects and contribute to vasodilation as a prevailing EDHF [98-100]. Watanabe and colleagues [86] determined that EETs stimulate TRPV4 channels in aortic ECs and may induce vasodilation. A subsequent study elucidated that exogenous 11,12-EET application in mesenteric arteries elicited vasodilation via hyperpolarization in VSMCs, and this vasomotor activity was substantially abrogated by TRPV4 knockout or the inhibition of Ca2+-sensitive K+ channels, but not by the blockade of NOS and COX [95]. More recently, pre-constricted human coronary arteries with endothelin-1 exhibited vasodilation in the presence of arachidonic acid, which was inhibited by RN-1734 [101]. In addition, the arachidonic acid-induced increase in Ca2+ concentration and vasodilation was profoundly reduced by the combination of TRAM-34 and apamin (inhibitors of IKCa and SKCa channels, respectively), the removal of extracellular Ca2+, or pretreatment with CYP inhibitors, suggesting that CYP-dependent metabolite generation from arachidonic acid increases Ca2+ influx (but not Ca2+ release from the sarcoplasmic reticulum) through EC TRPV4 channels and subsequently requires IKCa and SKCa-mediated hyperpolarization for vasodilation in human coronary arteries [101].
As most studies on TRPV4 channels have been performed under ex vivo preparations, many investigators have raised questions regarding the regulatory effect of TRPV4 channels on the cardiovascular system. The administration of GSK1016790A (a selective agonist for TRPV4 channels) interestingly decreased mean arterial pressure [102], which implies that the systemic activation of TRPV4 channels may contribute to the attenuation of peripheral resistance [32]. Consistently, there are studies showing that TRPV4-deficient mice did exhibit a higher mean arterial pressure compared with that of control mice [103]. Moreover, in the presence of the concurrent administration of an NOS inhibitor (L-NAME) and angiotensin II, TRPV4 knockout mice showed a much greater mean arterial pressure [104], indicating that TRPV4 may provide an alternative compensatory pathway in pathophysiological circumstances to prevent cardiovascular diseases (e.g., hypertension) by reducing peripheral resistance or inducing vasodilation [105].
TRPA1
TRPA1 channels are nonselective cation (i.e., Na+, Ca2+) channels (but specifically permeable to Ca2+ compared with Na+, PCa:PNa ≈ 7.9) that have a large unitary conductance (≈ 98 pS) [106,107]. As the name of the ion channel implies, TRPA1 channels have a distinguished structure that exhibits approximately 14–19 ankyrin repeat domains in the intracellular N-terminus of the ion channels [108]. The ankyrin repeat domains participate in protein-protein interactions and modulate channel activity in response to agonists or heat [109,110]. It is well established that diverse pungent dietary compounds such as allyl isothiocyanate (AITC) from mustard, allicin from garlic, eugenol from cloves, or cinnamaldehyde from cinnamon [111] activate TRPA1 channels by exerting covalent modifications on cysteine residues of the N-terminus of the channels [112,113]. Along with the dietary activators, oxidative stress and specific metabolites of lipid peroxidation act as physiological regulators for TRPA1 channels. Previous studies determined that endogenous aldehydes, 4-hydroxy-2-nonenal (4-HNE), 4-oxononenal (4-ONE), or 4-hydroxyhexenal (4-HHE) derived from the peroxidation of ω6 polyunsaturated fatty acids in the plasma membrane activate TRPA1 channels [114-118]. Further, hydrogen peroxide (H2O2) resulted in the oxidative modification of cysteine residues and subsequently activated TRPA1 channels in neuron cells [114,116]. Therefore, oxidative stress metabolites play a vital role in stimulating TRPA1 channels.
In light of the physiological consequences of TRPA1 activation, it was first noted that AITC application leads to vasorelaxation in rat mesenteric arteries by activating the target ion channels in perivascular nerves [119]. Interestingly, the AITC-mediated relaxant response was markedly attenuated after the pretreatment of the calcitonin gene-related peptide (CGRP) receptor antagonist (i.e., CGRP 8–37). Previous findings demonstrated that the dietary TRPA1 activators caused cultured trigeminal neurons to release CGRP, which was abolished by a TRPA1 channel blocker [120]. Therefore, it is likely that CGRP released from perivascular nerve endings elicits vasodilation. Subsequent studies further showed that cinnamaldehyde-mediated vasodilation in mesenteric arteries is considerably abrogated in TRPA1-deficient mice, whereas endothelial denudation has minor effects on vasodilation [121]. This suggests that pungent dietary activators of TRPA1 channels elicit vasodilation by inducing Ca2+ influx-dependent CGRP release from perivascular nerves and subsequently binding the peptide to CGRP receptors in VSMCs. Recently, it was reported that TRPA1 channels regulate the cold-induced vascular response [122]. Specifically, sequential vasoconstriction and vasodilation occur following local cold exposure to prevent cutaneous tissues from suffering from cold injury. Although TRPA1 channel-induced superoxide generation provoked vasoconstriction through α2-adrenoreceptor activation and myosin light chain phosphorylation immediately after environmental cold exposure, neuropeptide CGRP released from sensory nerves evoked TRPA1 channel-dependent vasodilation for the restoration of reduced blood flow [122].
Meanwhile, TRPA1 channels are identified in ECs and in perivascular or sensory nerves. However, the distribution of EC TRPA1 channels is limited to only the cerebral arteries, not the mesenteric, coronary, or renal arteries [28,37]. The direct influence of EC TRPA1 channels on the vasomotor response following the treatment of an electrophilic TRPA1 activator derived from mustard oil (e.g., AITC) was first assessed in pressurized cerebral arteries [37]. AITC concentration-dependent vasodilation of cerebral arteries was observed, which was accompanied by a significant reduction in the intracellular Ca2+ levels of cerebral arterial myocytes. Because VDCCs are a key determinant for Ca2+ dynamics in VSMCs, TRPA1 activation was suggested to induce VSMC hyperpolarization and vasodilation. TRPA1 channel-mediated vasodilation was diminished in EC-denuded arteries or after TRPA1 channel blocker (HC-030031) pretreatment [37], indicating that AITC-dependent vasodilation in cerebral arteries is EC-dependent and distinct from CGRP-induced vasorelaxation in mesenteric arteries [119]. Moreover, in intact ECs, TRPA1 channels are detected at MEPs where IKCa channels, but not SKCa channels, are locally expressed [37]. Pharmacological approaches have substantiated that although TRPA1-induced cerebral vasodilation is not affected by the inhibition of NOS and COX, the application of Ca2+-sensitive K+ channel blockers or a Kir channel inhibitor markedly suppresses AITC-induced vasodilation. Thus, EC TRPA1-dependent Ca2+ entry (called TRPA1 Ca2+ sparklets) elicits a K+ efflux via IKCa/SKCa channels and provokes vasodilation through EC and VSMC hyperpolarization, which is amplified by Kir channels in cerebral arteriolar myocytes [37]. Earley and co-workers extended their novel hypotheses on TRPA1 channel-induced vasodilation and revealed innovative cerebral dilatory mechanisms. They found that EC TRPA1 channels frequently display a binary channel gating behavior (i.e., two TRPA1 channels open simultaneously) [28]. Their study focused on the ROS-mediated activation of TRPA1 channels in cerebral arteries [28]. NADPH (nicotinamide adenine dinucleotide phosphate) oxidase (NOX) was shown to generate ROS [123,124], and TRPA1 channels in neurons are stimulated by ROS or ROS-derived metabolites [114,116,125]. In cerebral arteries, the administration of exogenous NADPH-induced vasodilation that was profoundly decreased in the presence of HC-030031 or TRAM-34 [28]. Consistent with their prior study [37], the combination of L-NNA and indomethacin did not affect NADPH-induced vasodilation [28]. Collectively, NADPH-NOX downstream signaling was shown to stimulate Ca2+ entry via EC TRPA1 channels and subsequently led to IKCa channel-dependent vasodilation. To investigate specific signaling pathways, diverse pharmacological interventions including apocynin (an NOX inhibitor), catalase (a hydrogen peroxide-metabolizing enzyme), and deferoxamine (an inhibitor for the hydroxyl radical and lipid peroxidation) were employed in that study [28]. As a result, NADPH-mediated vasodilation was completely abolished in the presence of those inhibitors, suggesting that NOX-derived H2O2 causes the production of lipid peroxidation metabolites, which stimulate EC TRPA1 Ca2+ sparklets and provoke IKCa activation-dependent vasodilation in cerebral arteries [28]. Therefore, since NOX-derived ROS production was reported to be much greater in the cerebral vasculature than in other vascular beds [126], it is strongly noted that ROS and lipid peroxidation products may serve as endogenous modulators of TRPA1 channels and physiological regulators for vasomotor reactivity in the cerebral circulation.
Go to :
CONCLUSIONS AND PERSPECTIVES
Impaired endothelial function has been documented as a hallmark of cardiovascular disorders including hypertension, vasospasm, atherosclerosis, and coronary/cerebral ischemia. The endothelium plays an important role in the modulation of vascular contractility, which is regulated by alterations in endothelial Ca2+ signaling. Despite the significance of Ca2+ dynamics in ECs, the mechanisms underlying endothelium-dependent Ca2+ influx and hyperpolarization remain uncertain. In light of this, TRP channels have drawn attention to the regulation of Ca2+ mobilization and vascular tone via EDH-mediated vasorelaxation/dilation, as have advances in optical Ca2+ imaging techniques and highly sensitive Ca2+-detecting molecules (e.g., fluo-4, GCaMP2 biosensor). As a result, previous groundbreaking studies have demonstrated that TRP channels participate in Ca2+ entry and in turn induce EDH-mediated vasodilation in small resistance arteries. However, because most of those works have utilized pharmacological agonists/antagonists to explore physiological outcomes and biophysical properties of the TRP channels under ex vivo preparation, the endogenous modulators for TRP channel-dependent endothelial Ca2+ signaling must be identified and the effects of biological stimuli (e.g., shear stress, intravascular pressure, stretch/tension of plasma membrane) on the activation of TRP channels in the ECs of resistance arteries must be studied. It is strongly believed that such studies will contribute to an exact understanding of the complexity of EC Ca2+ signaling through TRP channels and the discovery of therapeutic targets for cardiovascular disease treatments.
Go to :
 XML Download
XML Download