

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Polycystic kidney disease 2-like-1 (PKD2L1) is known to modulate ciliary calcium concentration and has recently been reported to be involved in mechanoception in neurons [1,2]. PKD2L1 forms a functional complex with PKD1 homologs, PKD1L1 and PKD1L3, and regulates hedgehog pathways and sour sensation, respectively [3-5]. PKD2L1 has been known to be regulated in response to extracellular and intracellular calcium concentrations [6]. In our previous study, we identified how PKD2L1 channel activation is regulated by the cyclic adenosine monophosphate (cAMP) signaling pathway by identifying the clustered phosphorylation site of PKD2L1 [7]. The structure of PKD2L1 has also been reported [8,9], but further studies on the functional role of C-terminus of the channel, including potential calmodulin-binding domain (CaMBD), are needed.
Calmodulin consists of two lobes, N-lobe and C-lobe, and by binding calcium with EF-hands, it results in conformational change, signaling to various targets [10,11]. Although the two lobes show a high sequence identity, the C-lobe has higher calcium affinity than the N-lobe [12,13]. This leads to subtle differences in target recognition [14] and consequently plays an important role in CaM function. CaM is a calcium binding protein and is well known as an ion channel activity regulator [15,16]. CaM has two effects, Ca2+-dependent facilitation (CDF) and Ca2+-dependent inhibition (CDI), depending on the targeted ion channel [17,18]. These two effects are caused by various interactions with CaM such as CaMBD and IQ motif of ion channel.
In small-conductance Ca2+-activated K+ (SK) channels, the C-lobe of CaM remains attached to the channel, and N-lobe is known to be related to the gating mechanism by interacting with the S4-S5 linker depending on calcium [19]. The voltage-gated Na+ (NaV) channel also depends on calcium and binds to CaM at the C-terminus [20]. The voltage-gated sodium channel NaV1.5 (hH1) causes a molecular switch that attenuates the interaction between CaM and IQ and transforms it into binding to EF-hand by calcium signal [21]. Voltage-gated Ca2+ (CaV) channels were known to form its own complex with the IQ domain at the C-terminus of the channel, but at CaV1.3 it was reported that CaM N-lobe binds to the N-terminus and C-lobe binds to the EF-hand of the channel [22].
Transient receptor potential (TRP) channels with calcium permeability perform negative feedback by calcium permeation to maintain calcium homeostasis, and kinase, phosphatase, phospholipase and CaM are the causes of calcium-dependent desensitization [23]. TRP ankyrin 1 (TRPA1) binds to CaM at the C-terminus and regulates its sensitization according to calcium concentration [24]. TRP canonical (TRPC) channels have multiple CaM-binding sites, and at the C-terminus of all TRPC isoforms, there is a CaM/inositol 1,4,5-trisphosphate receptor-binding (CIRB) site and an additional non-conserved CaM-binding site [25]. The coiled-coil assembly of TRPC6 channels is involved in CDI, and defects in this process are related to focal segmental glomerulosclerosis (FSGS) [26]. TRP vanilloid 5 (TRPV5) has also reported a mechanism by which CaM depends on calcium to regulate channels and maintain calcium homeostasis [27]. In TRPV6, the mechanism by which CDI occurs when the tetramer of TRPV6 binds to two lobes of CaM has been described [28]. CaM in PKD2L1 delayed channel potentiation time course by inhibiting channel activity, and N-lobe has been reported to play a key role in regulating PKD2L1 [29].
There are various structural ways in which CaM recognizes and complexes multiple targets, including ion channels [30]. Structural features and commonalities have been discovered through these various CaM-complex structures, and several canonical CaM-binding motifs are known [31,32]. The canonical CaM-binding motifs have several motifs, depending on the number of amino acid residues between the hydrophobic anchor residues. This hydrophobic anchor residue is [FILVWY], and it is often replaced by a different type of residue depending on calcium dependence and type of channel, so there is no defined CaM-binding recognition sequence. The CaM antagonist calmidazolium (CMZ) is also known as an activator of the PKD2L1 channel, and the activation mechanism is not yet known. CMZ has a nonspecific effect that can cause pharmacological effects by blocking L-type Ca2+, K+, Na+ channels and sarcoplasmic reticulum (SR) calcium release channels [33,34].
CaM is known to bind to a variety of targets, including CaMBD of ion channels, but the binding site of CaM in PKD2L1 binding to CaM has not been identified. Here, to confirm the putative CaMBD (K590-E600) expected in the previous study, single mutants of this region and EF-hand deleted mutants at the same time were constructed and recorded with CMZ treatment under different intracellular calcium concentrations using patch-clamp technique. Overall, this study suggests a mechanism by which CaM regulates PKD2L1 channel activation.
METHODS
Cell culture and transient transfection
Human embryonic kidney 293 (HEK293) cells (American Type Culture Collection, Manassas, VA, USA) were cultured according to the supplier’s recommendations. For transient transfection, the cells were seeded in 12-well plates. The following day, 0.5 μg/well of internal ribosome entry site (IRES) and enhanced green fluorescence protein (EGFP) containing human PKD2L1 was transfected into cells using FuGENE 6 transfection reagent (Promega, Madison, WI, USA) according to the manufacturer’s protocol. For co-transfection with CaM, 1 μg/well was transfected into cells with human PKD2L1-IRES-EGFP at 2:1. Within 24 to 48 h, the attached cells were trypsinized and used for whole-cell recording.
Molecular biology
Point mutations in human PKD2L1 were performed by QuickChange site-directed mutagenesis kit (Agilent Technologies, Santa Clara, CA, USA). Sequences of all constructs were confirmed by DNA sequencing.
Electrophysiology
Whole cell currents were recorded using an Axopatch 200B amplifier (Axon Instruments, Foster City, CA, USA). Currents were filtered at 5 kHz (–3 dB, 4-pole bessel), digitized using a Digidata 1440A Interface (Axon Instruments), and analyzed using a personal computer equipped with pClamp 10.2 software (Axon Instruments) and Origin software (Microcal origin v.8.0; Microcal Software, Northampton, MA, USA). Glass microelectrodes had resistances 2 to 4 MΩ when filled with internal solution. From holding potential at –60 mV, voltage ramp pulse was applied from –100 mV to 100 mV for 500 ms. For whole cell experiments, we used an external bath medium (standard bath solution) of the following composition (in mM): 150 NaCl, 10 N-(2-hydroxyethyl)piperazine-N’-2-ethansulfonic acid (HEPES), 1.8 CaCl2, and 1 MgCl2 with pH adjusted to 7.4 using NaOH. The internal solutions contained (in mM): 100 CsMES, 35 NaCl, 10 HEPES, 5 cesium-1,2-bis-(2-aminophenoxy) ethane-N,N,N’,N’-tetraacetic acid (Cs-BAPTA), 2 MgCl2, 1.058 CaCl2 for 16 nM free Ca2+, 2.581 CaCl2 for 100 nM free Ca2+, with pH adjusted to 7.4 using CsOH. For recording the current responses of the channels to CaM inhibitors, 1 μM of calmidazolium (CMZ) (Tocris Bioscience, Bristol, United Kingdom) was used dissolved in standard bath solution (SBS). The diluted solution was made on each day of experiment and any remainder of the solution was discarded after the experiment. Experiments were performed at room temperature (20°C–24°C).
Statistics
Statistical analysis was done using GraphPad Prism 5 (GraphPad Software Inc., San Diego, CA, USA). Results are presented as means ± standard error of mean. Statistical data were compared by paired or unpaired Student’s t-test between two groups. The p-values less than 0.05 were considered statistically significant. The number of whole cell recordings is indicated by n.
RESULTS
Prediction of PKD2L1 putative CaM-binding Domain
In our previous study, we used the Calmodulin Target Database to predict the CaMBD of PKD2L1 [29]. As a result, the protein sequence between K590 and E600 of PKD2L1 showed a high score of 7 to 9, indicating a very important site for CaM binding. The protein sequence of the putative CaMBD (K590-E600) of PKD2L1 is contained within the oligomerization domain (OD) (Fig. 1). Based on this, to identify CaM-binding anchor residue of PKD2L1, single mutants were prepared by substituting alanine for each amino acid of this domain.
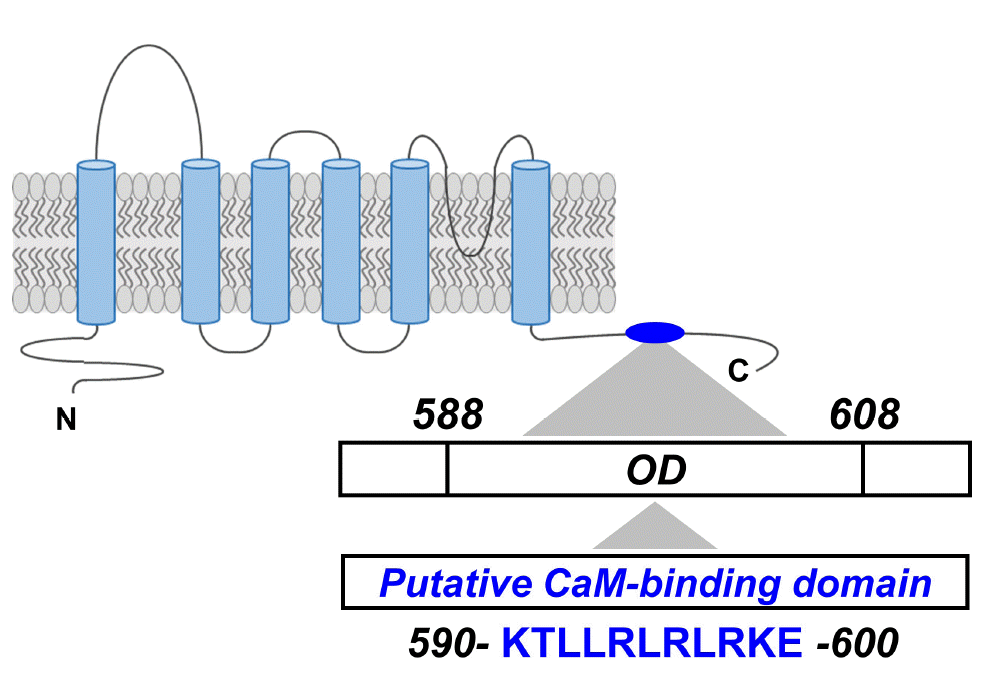
Identification of putative CaM-binding domain (K590-E600) in PKD2L1
We have recently confirmed different time courses of PKD2L1 channel potentiation and inactivation according to different intracellular calcium concentrations [29]. To determine the regulation of PKD2L1 channel activity of CaM at different intracellular calcium concentrations, we used 16 nM as below normal calcium and 100 nM as normal intracellular calcium concentrations. First we confirmed whether the effect of CMZ, the antagonist of CaM and the activator of PKD2L1, depends on calcium condition. Application of 1 μM CMZ on the PKD2L1-expressed HEK293 cell increased the currents from 45 ± 4 pA/pF (n = 32) to 96 ± 6 pA/pF (n = 32) under 16 nM free Ca2+ and with 100 nM free Ca2+, from 39 ± 8 pA/pF (n = 17) to 93 ± 10 pA/pF (n = 17) (Fig. 2).
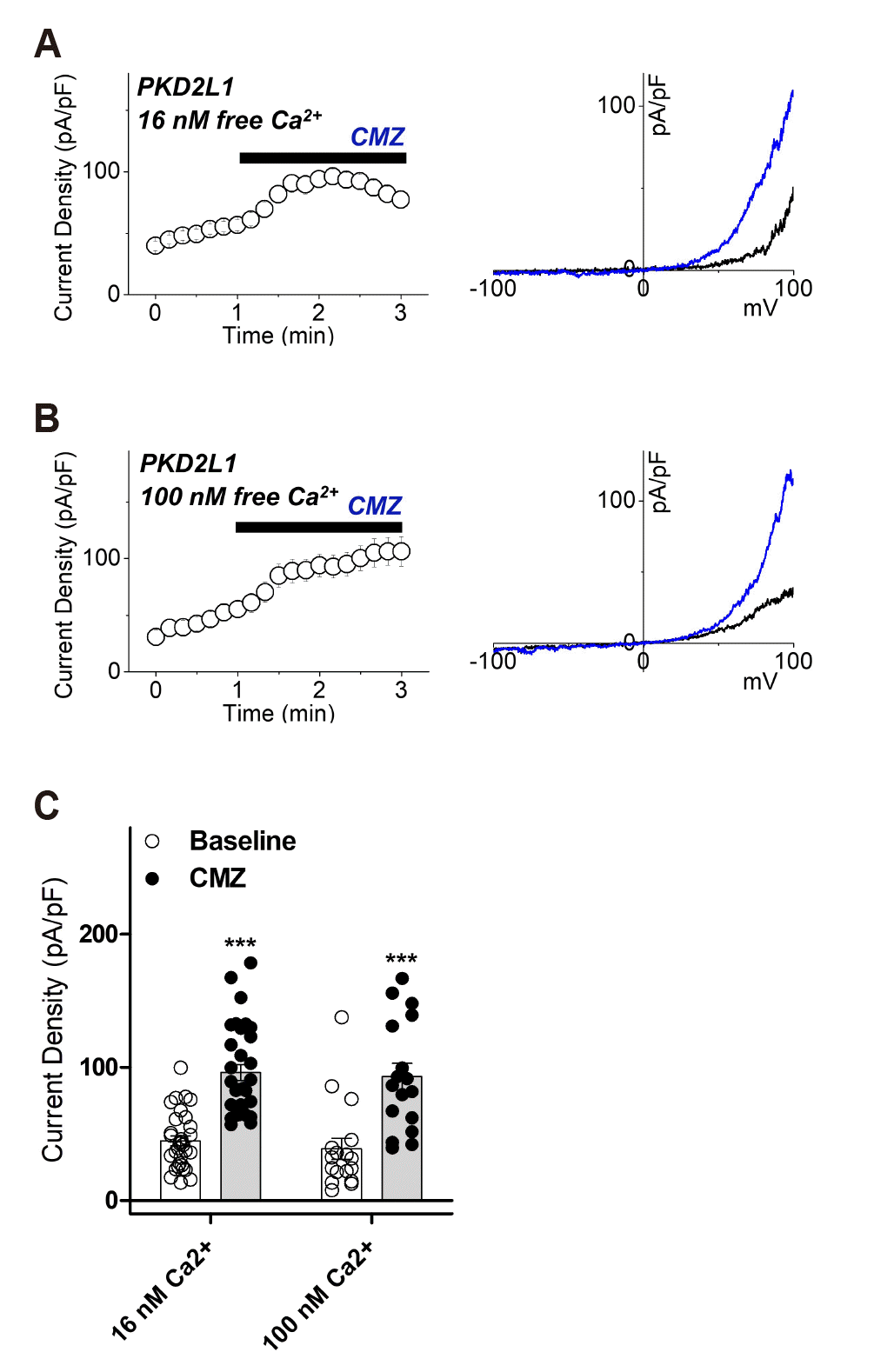
Fig. 2
The effects of calmidazoluim (CMZ) on polycystic kidney disease 2-like-1 (PKD2L1) current under 16 nM and 100 nM intracellular free calcium concentrations.
(A) A full current trace of PKD2L1 (left) activated by 1 μM of CMZ under 16 nM free Ca2+ and the current (I)–voltage (V) relationship of PKD2L1 (right) at the basal current amplitude (black) and at the application of 1 μM of CMZ (blue). (B) A full current trace of PKD2L1 (left) activated by 1 μM of CMZ under 100 nM free Ca2+ and the I–V relationship of PKD2L1 (right) at the basal current amplitude (black) and at the application of 1 μM of CMZ (blue). (C) A summarized current amplitude of PKD2L1 induced by CMZ under 16 nM and 100 nM free Ca2+ (n = 17–32). ***p < 0.001.
![]()
The currents of single mutants in the putative CaMBD (K590-E600) were recorded in 1 μM CMZ stimulation under 16 nM and 100 nM free calcium conditions. If the mutant is indeed a CaM-binding site, CaM would not bind to PKD2L1, and it is expected that there would be little current change by CMZ. Each mutant showed various differences in comparison with the basal current of PKD2L1 wild type as well as various responses to CMZ, and their response patterns could be classified into three types. First, there were mutants that are likely to lose their activity as channel because the basal current is significantly lower than the PKD2L1 wild type. Under 16 nM free Ca2+, as shown in Fig. 3D, K590A, R596A, R598A, K599A and E600A mutants showed a basal current significantly lower than that of the PKD2L1 wild type, exhibiting reduction in channel activity to 7 ± 2 pA/pF (n = 4), 11 ± 1 pA/pF (n = 7), 15 ± 3 pA/pF (n = 4), 11 ± 3 pA/pF (n = 5) and 9 ± 3 pA/pF (n = 4), respectively. Second, among those with basal current similar to or higher than that of the PKD2L1 wild type, there were mutants that responded to CMZ. L592A and R594 were significantly higher than the PKD2L1 wild type basal current, and the CMZ-induced current changes were 100 ± 39 pA/pF (n = 5) and 132 ± 51 pA/pF (n = 4), respectively, which were significantly higher than the wild type (Fig. 3D, F). Third, among the mutants whose basal current is similar to or higher than that of the PKD2L1 wild type, there were mutants whose current change by CMZ was similar to the wild type. T591A, L593A, L595A, and L597A had basal currents similar to those of the PKD2L1 wild type, which showed 66 ± 12 pA/pF (n = 6), 60 ± 24 pA/pF (n = 6), 61 ± 23 pA/pF (n = 5), and 56 ± 22 pA/pF (n = 5) due to the change in current caused by CMZ, which was similar to the wild type (Fig. 3D, F). As a result of the CMZ treatment of the putative CaMBD (K590-E600) single mutants under 16 nM intracellular calcium concentration, Thr-591, Leu-593, Leu-595 and Leu-597 were selected as candidates for the putative CaM-binding anchor residue.
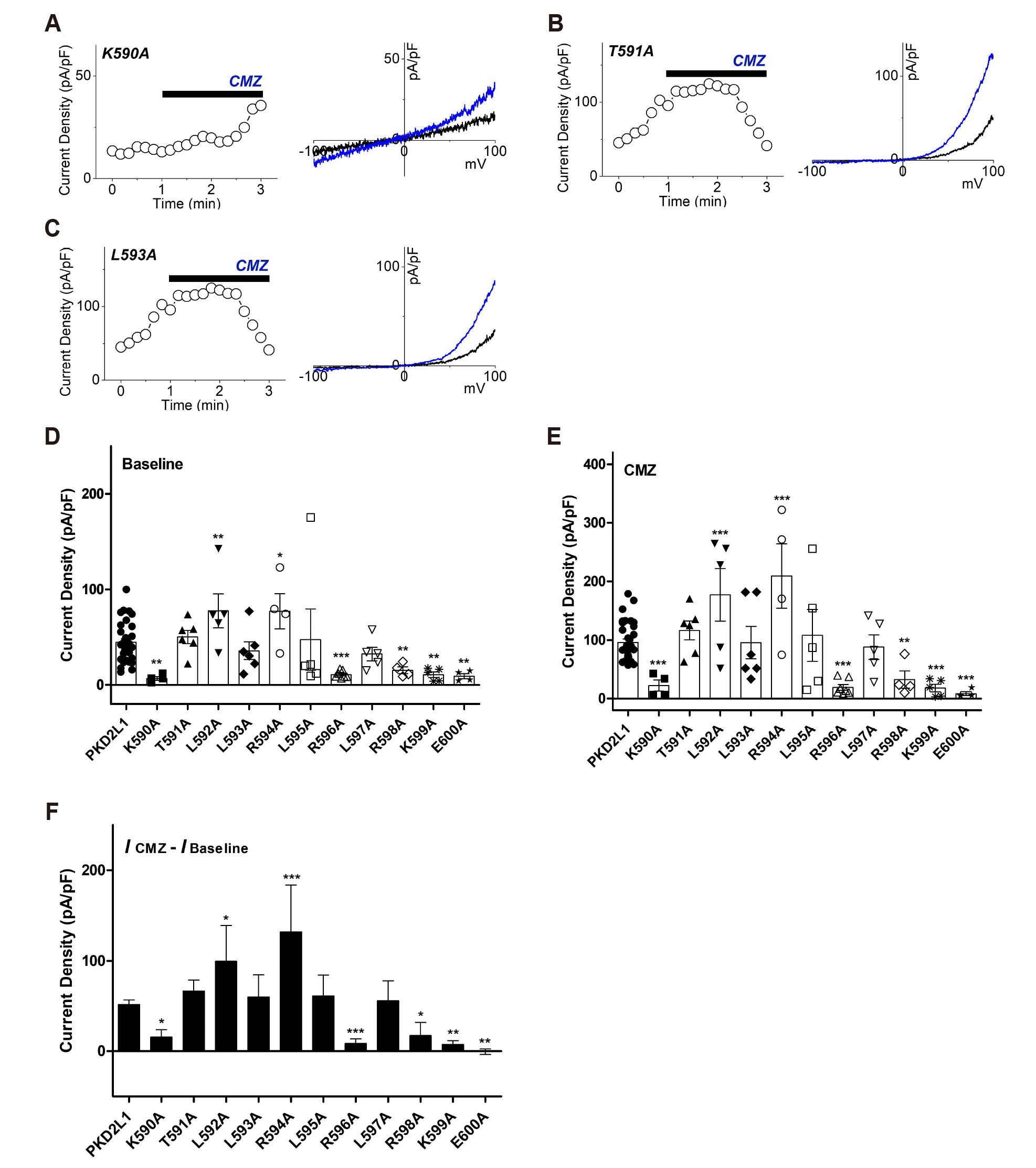
Fig. 3
The effects of calmidazolium (CMZ) on putative calmodulin-binding domain (CaMBD) (K590-E600) single mutants under 16 nM intracellular free calcium concentration.
(A) A full current trace (left) and the current (I)–voltage (V) relationship (right) of polycystic kidney disease 2-like-1 (PKD2L1) (K590A) activated by 1 μM of CMZ (blue) under 16 nM free Ca2+. (B) A full current trace (left) and the I–V relationship (right) of PKD2L1 (T591A) activated by 1 μM of CMZ (blue) under 16 nM free Ca2+. (C) A full current trace (left) and the I–V relationship (right) of PKD2L1 (L593A) activated by 1 μM of CMZ (blue) under 16 nM free Ca2+. (D) A summarized basal current amplitude of PKD2L1 and single mutants under 16 nM free Ca2+ (n = 4–32). (E) A summarized CMZ-induced current amplitude of PKD2L1 and single mutants under 16 nM free Ca2+ (n = 4–32). (F) A summarized current changes of PKD2L1 and single mutants by CMZ under 16 nM free Ca2+. *p < 0.05, **p < 0.01, ***p < 0.001.
![]()
Identification of putative CaM-binding domain (K590-E600) in PKD2L1 with EF-hand deleted
To determine if EF-hand affects the binding of CaM to PKD2L1, we first identified whether there is a change in current by the CMZ in EF-hand deletion mutant (ΔEF) under 16 and 100 nM free calcium conditions. When 1 μM CMZ was applied, the current of ΔEF mutant increased from 41 ± 10 pA/pF (n = 4) to 124 ± 26 pA/pF (n = 4) under 16 nM free Ca2+ and with 100 nM free Ca2+, from 47 ± 12 pA/pF (n = 5) to 167 ± 17 pA/pF (n = 5) (Fig. 4).
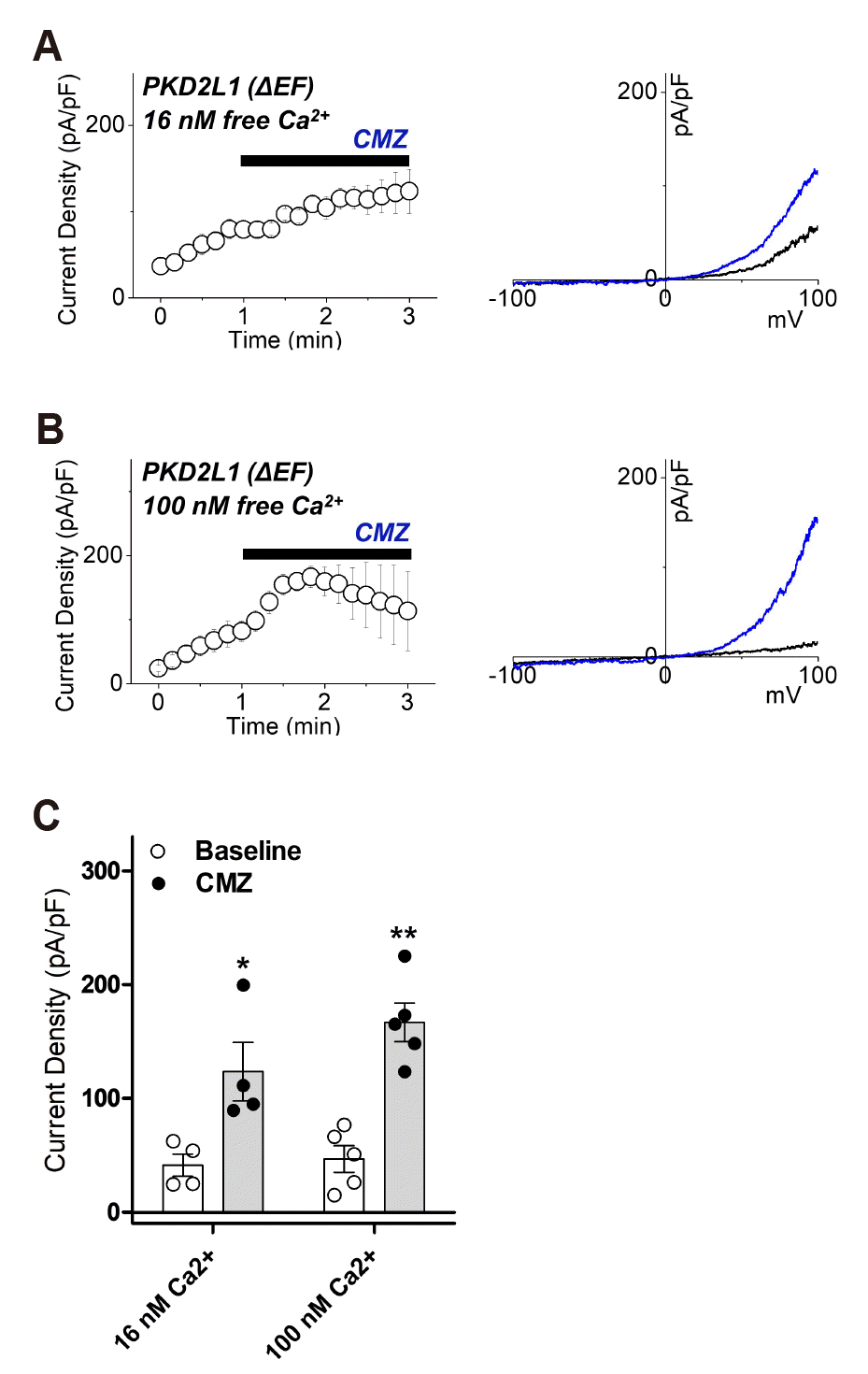
Fig. 4
The effects of calmidazolium (CMZ) on polycystic kidney disease 2-like-1 (PKD2L1) ΔEF current under 16 nM and 100 nM intracellular free calcium concentrations.
(A) A full current trace of PKD2L1 (ΔEF) (left) activated by 1 μM of CMZ under 16 nM free Ca2+ and the current (I)–voltage (V) relationship of PKD2L1 (ΔEF) (right) at the basal current amplitude (black) and at the application of 1 μM of CMZ (blue). (B) A full current trace of PKD2L1 (ΔEF) (left) activated by 1 μM of CMZ under 100 nM free Ca2+ and the I–V relationship of PKD2L1 ΔEF (right) at the basal current amplitude (black) and at the application of 1 μM of CMZ (blue). (C) A summarized current amplitude of PKD2L1 (ΔEF) induced by CMZ under 16 nM and 100 nM free Ca2+ (n = 4–5). *p < 0.05, **p < 0.01.
![]()
Next, mutants of CaMBD (K590-E600), in which EF-hand was deleted, were prepared to confirm the current change in 16 nM free Ca2+ and 100 nM free Ca2+. At 16 nM free Ca2+, the response of the mutants was categorized into two. First, as shown in Fig. 5D, K590/ΔEF, L595A/ΔEF and L597A/ΔEF showed reduction in channel activity to 13 ± 3 pA/pF (n = 4), 14 ± 6 pA/pF (n = 4) and 5 ± 1 pA/pF (n = 4), respectively. The rest of the group is mutants with similar basal currents to the wild type, at the same time the current change due to CMZ is no different or lower than the wild type. T591A/ΔEF, L593A/ΔEF, R594A/ΔEF, R596A/ΔEF, R598A/ΔEF, K599A/ΔEF and E600A/ΔEF have a basal current similar to wild type, and 113 ± 18 pA/pF (n = 4), 60 ± 23 pA/pF (n = 4), 85 ± 28 pA/pF (n = 4), 99 ± 47 pA/pF (n = 4), 52 ± 15 pA/pF (n = 6), 98 ± 21 pA/pF (n = 4), 91 ± 11 pA/pF (n = 5) showed a range of current change by CMZ induction similar to wild type (Fig. 5D, F). The L592A/ΔEF had a basal current similar to that of the wild type, and the range of current change induced by CMZ was 11 ± 6 pA/pF (n = 5), which is lower than that of the wild type (Fig. 5D, F). CMZ treatment of the putative CaMBD (K590-E600) mutants with EF-hand deletion under 16 nM intracellular calcium concentration resulted in Thr-591, Leu-592, Leu-593, Arg-594, Arg-596, Arg-598, Lys-599 and Glu-600 showed the potential as CaM-binding anchor residue. From the results in Figs. 3 and 5, Thr-591 and Leu-593 could be deduced with the possibility of a common CaM-binding anchor residue at 16 nM calcium levels irrespective of EF-hand.
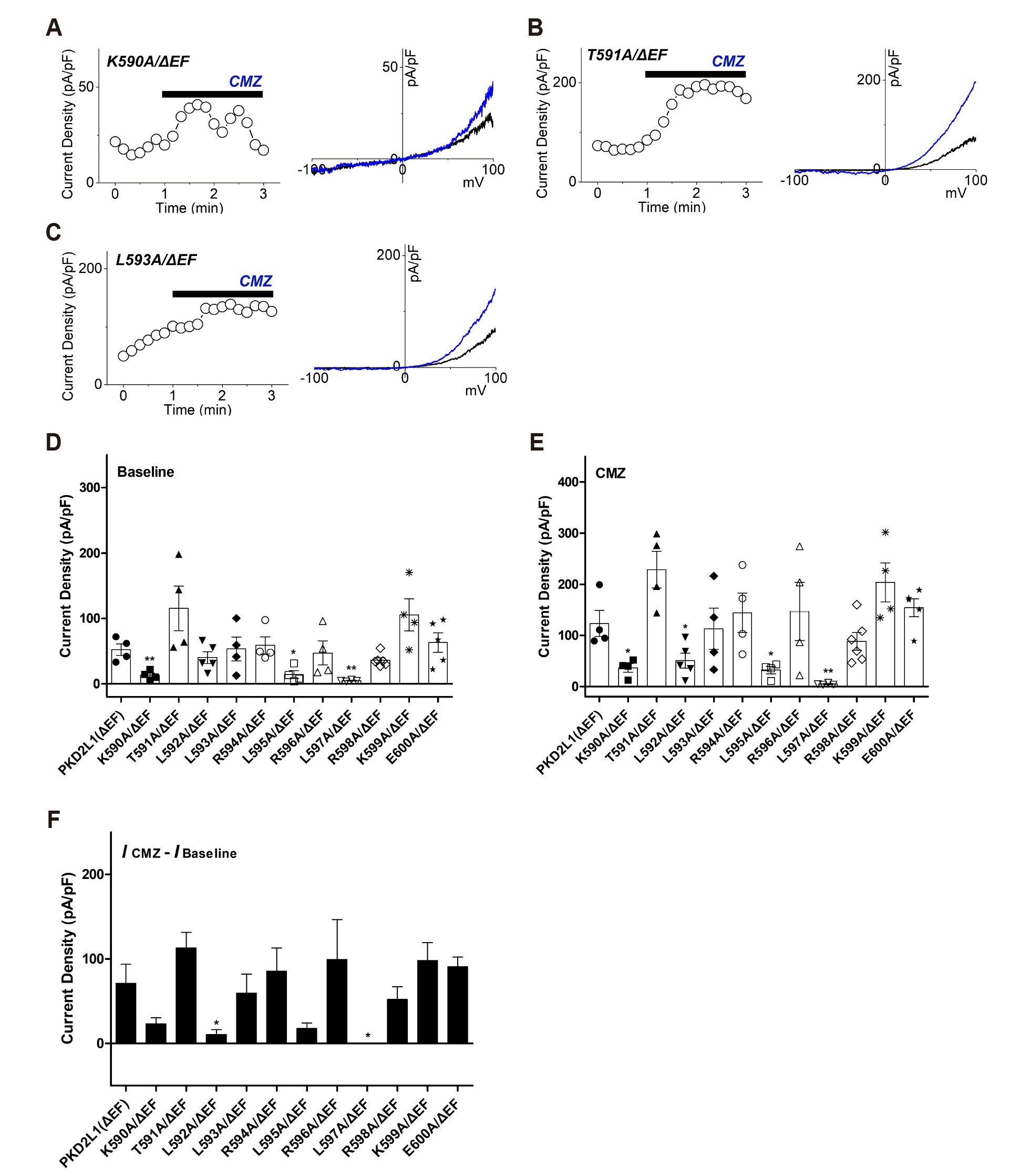
Fig. 5
The effects of calmidazolium (CMZ) on putative calmodulin-binding domain (CaMBD) (K590-E600)/ΔEF mutants under 16 nM intracellular free calcium concentration.
(A) A full current trace (left) and the current (I)–voltage (V) relationship (right) of polycystic kidney disease 2-like-1 (PKD2L1) (K590A/ΔEF) activated by 1 μM of CMZ (blue) under 16 nM free Ca2+. (B) A full current trace (left) and the I–V relationship (right) of PKD2L1 (T591A/ΔEF) activated by 1 μM of CMZ (blue) under 16 nM free Ca2+. (C) A full current trace (left) and the I–V relationship (right) of PKD2L1 (L593A/ΔEF) activated by 1 μM of CMZ (blue) under 16 nM free Ca2+. (D) A summarized basal current amplitude of PKD2L1 (ΔEF) and mutants under 16 nM free Ca2+ (n = 4–6). (E) A summarized CMZ-induced current amplitude of PKD2L1 (ΔEF) and mutants under 16 nM free Ca2+ (n = 4–6). (F) A summarized current changes of PKD2L1 (ΔEF) and mutants by CMZ under 16 nM free Ca2+. *p < 0.05, **p < 0.01.
![]()
PKD2L1 Leu-593 has potential as CaM C-lobe anchor residue
Additionally, current changes by CMZ of single mutants of the putative CaMBD (K590-E600) and mutants simultaneously deleted with EF-hand under 100 nM calcium concentration were also recorded. CMZ treatment of the putative CaMBD (K590-E600) single mutants under 100 nM intracellular calcium concentration showed that the basal current of all single mutants, K590A, T591A, L592A, L593A, R594A, L595A, R596A, L597A, R598A, K599A and E600A, were similar to that of the wild type, and CMZ-induced current change was less than or similar to wild type (Supplementary Fig. 1). CMZ treatment of the putative CaMBD (K590-E600) mutants with EF-hand deletion at 100 nM free calcium showed that basal current of T591A/ΔEF, L592A/ΔEF, R594A/ΔEF, R596A/ΔEF, R598A/ΔEF and E600A/ΔEF were similar or high compared with wild type and the CMZ-induced currents did not show significant difference from that of the wild type (Supplementary Fig. 2). CMZ-treated current changes of the putative CaMBD (K590-E600) single mutants under 100 nM free calcium showed that all single mutants have potential as CaM-binding anchor residue. For this reason, PKD2L1 has its potentiation at 100 nM calcium [29]. Therefore, it was difficult to distinguish the effect of CMZ at 100 nM Ca2+. The deletion of PKD2L1 EF-hand at 100 nM calcium is negligible when current changes are compared with wild type [29]. As a result, the current change of PKD2L1 ΔEF at 100 nM calcium was similar to the wild type current change. However, the change of CMZ current could not be determined because of the potentiation of PKD2Ll under 100 nM calcium condition.
T591A was excluded because it was identified as a nonspecific site regardless of intracellular calcium concentration and EF-hand. In conclusion, Leu-593 was considered as a CaM-binding anchor residue. According to the Calmodulin Target Database of hPKD2L1, Leu-593 has a score of 9 points, which was analyzed as a highly relevant site for CaM-binding [29]. Under the 16 nM calcium concentration, the basal current of K590A was significantly lower than the wild type. The mutant exhibiting functional deficiencies suggests that the site is crucial for maintaining channel activity. Western blot analysis showed that the expression level of mutants deleted from putative CaMBD (K590-E600) was not significantly different from that of PKD2L1 wild type [29].
Analysis of the above results confirmed that Leu-593 is a possible CaM-binding anchor residue. CaM N-lobe has been reported to play a more important role than C-lobe in inhibiting PKD2L1 channel activation [29]. In addition, it was expected that CaM N-lobe bound to PKD2L1 at 100 nM calcium because CaM inhibited more PKD2L1 at 100 nM calcium than at 16 nM calcium. In this study, Leu-593 of PKD2L1 was predicted as CaM C-lobe anchor residue because it was analyzed under 16 nM calcium concentration.
Next, to confirm that Leu-593 is the anchor residue, we examined whether there is a current change when L593A is co-expressed with CaM. At 16 nM free Ca2+, L593A had a peak current of 52 ± 21 pA/pF (n = 4) and 20 ± 5 pA/pF (n = 4) when co-expressed with CaM, showing no significant difference (Fig. 6A, C). On the other hand, when co-expressed with CaM at 100 nM free Ca2+, the peak current of L593A reduced significantly from 159 ± 32 pA/pF (n = 4) to 42 ± 13 pA/pF (n = 4) (Fig. 6B, C). The potentiation time of L593A in both 16 and 100 nM free Ca2+ showed no significant change compared to the co-expression with CaM, yielding from 305 ± 52 sec (n = 4) to 358 ± 37 sec (n = 4) and from 358 ± 17 sec (n = 4) to 380 ± 33 sec (n = 4), respectively (Fig. 6D).
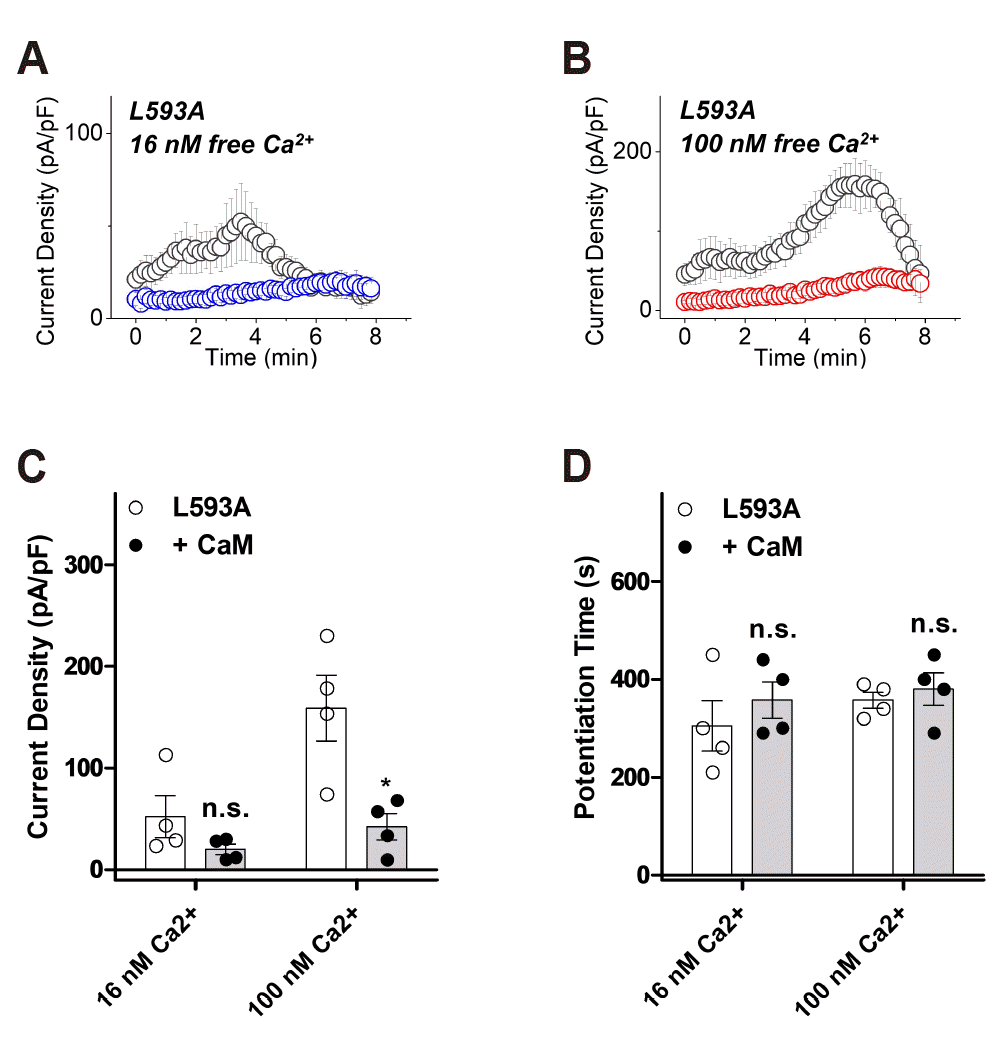
Fig. 6
The potentiation and inactivation of polycystic kidney disease 2-like-1 (PKD2L1) (L593A) with over-expression of calmodulin (CaM) under 16 nM and 100 nM intracellular free calcium concentrations.
(A) A full current trace of PKD2L1 (L593A) (gray) and the mutant co-expressed with CaM (blue) under 16 nM free Ca2+. (B) A full current trace of PKD2L1 (L593A) (gray) and the mutant co-expressed with CaM (red) under 100 nM free Ca2+. (C) A summarized peak current amplitude of PKD2L1 (L593A) and the mutant co-expressed with CaM under 16 nM and 100 nM free Ca2+ (n = 4). (D) A summarized peak time of PKD2L1 (L593A) and the mutant co-expressed with CaM under 16 nM and 100 nM free Ca2+ (n = 4). *p < 0.05.
![]()
DISCUSSION
CaM is known to bind to various targets, including the CaMBD in the channel, but the CaMBD and its inhibitory mechanism of PKD2L1 have not yet been identified. To investigate putative CaMBD (K590-E600) of PKD2L1 that we previously predicted (Fig. 1), we generated single mutants of this domain and also made double mutants with EF-hand deletion. The current changes were recorded using CMZ at calcium concentrations in 16 nM and 100 nM. CMZ is known as an antagonist of CaM and an activator of PKD2L1. In the case of mutants that mutated the CaM-binding site, CaM could not bind to this site, and the current change caused by CMZ expected to be minimal. As a result of patch-clamp, each mutant showed various differences in comparison with basal current of PKD2L1 wild type as well as various responses to CMZ. Based on basal current and CMZ current change, it could be classified into three groups. First, there are mutants that show complete loss of channel activity due to significantly lower basal current compared to the PKD2L1 wild type. Second, there are mutants that show a significant increase in current change due to CMZ, among those with similar or higher basal currents compared to the PKD2L1 wild type. The last group is the group of mutants that showed similar or higher basal currents compared with that of the wild type and no change in currents by CMZ treatment. They are highly likely to be the CaM-binding anchor residues. Based on the current change induced by CMZ, we speculated that Leu-593 is a possible CaM C-lobe anchor residue. The co-expression of L593A, a mutant form of Leu-593, with CaM, confirmed that the inhibition of PKD2L1 activity by CaM was weakened (Fig. 6). This again supports the possibility of Leu-593 as a CaM C-lobe anchor residue. In 100 nM calcium, the inhibitory effect of CaM on PKD2L1 remains (Fig. 6B, C). The reason for this is the possibility that CaM N-lobe binds to PKD2L1 and regulates channel activity irrespective of C-lobe and the presence of CaM-binding site in addition to the C-terminal domain of PKD2L1.
In addition, we identified the amino acid sequence of putative CaMBD in PKD2L1 by aligning it with rTRPV1 (Transient receptor potential vanilloid1). The CaM-binding motif and the CaM-binding anchor residue of rTRPV1 channel have been studied and reported. The matching sequence is shaded in gray, the putative CaMBD of PKD2L1 is underlined, and the CaM-binding anchor residues are marked in red (Supplementary Fig. 3). Leu-592, Leu-593 and Arg-594 of PKD2L1 were consistent with Leu-795, Leu-796 and Arg-797 of rTRPV1. Canonical CaM-binding motifs are known to be defined by varying spacing depending on the number of amino acids between the hydrophobic anchor residue [FILVWY]. The binding to CaM appears to play a large role in the anchor residue of the target [31,32]. rTRPV1 has two CaM-binding sites, N-terminal ankyrin repeat domain (ARD) and C-terminal domain (CTD) [35]. Trp-787 and Leu-796 of rTRPV1 are known as CaM C-lobe and N-lobe hydrophobic anchor residues, respectively, and their structure is similar to the canonical calcium-peptide complex, but rTPRV1 does not have a protein sequence that recognizes typical CaM. In addition, extending from the putative CaMBD of PKD2L1, PKD2L1 Leu-584 was composed of hydrophobic residue like rTRPV1 Trp-787. This predicts that the complex of PKD2L1 and CaM may be related to 1-10 motif, the CaM-binding motif of rTRPV1, which requires further study. Rat TRPV1 Trp-787 and Leu-796 were identified as conserved sequences in mouse and human TRPV1, and human PKD2L1 Leu-593 was also identified as a conserved sequence in mouse PKD2L1, which is expected to retain conserved functions that do not change between species. As a result of comparison with CaMBD of TRPV1, it was confirmed that Leu-593 of PKD2L1 is highly likely to play a role as CaM C-lobe anchor residue.
Based on our data, we predicted the mechanism by which CaM regulates PKD2L1 as shown in the following model (Fig. 7). It is expected that PKD2L1 EF-hand and CaM N-lobe will compete in channel binding because PKD2L1 EF-hand shows a protein sequence similar to that of CaM EF-2 included in CaM N-lobe. At 16 nM calcium concentration, EF-hand shows a persistent binding to PKD2L1, demonstrating its self-inhibition of channel activity. CaM C-lobe binds to PKD2L1 via CaM C-lobe anchor residue Leu-593 (Fig. 7A, upper panel). When EF-hand is deleted, the self-inhibition of the channel by coupling with EF-hand is weakened (Fig. 7A, lower panel). When the calcium concentration increases to 100 nM in a steady state, calcium first binds to CaM C-lobe, which has relatively high affinity. CaM N-lobe is then placed in a position to bind to the channel instead of EF-hand, which strongly inhibits the activity of PKD2L1 (Fig. 7B, upper panel). When the EF-hand is deleted, CaM N-lobe is already occupied at the position where EF-hand can bind, which still shows strong inhibition of the channel. At 100 nM, the role of the EF-hand channel is negligible (Fig. 7B, lower panel). In this study, we suggested that Leu-593 serves as the CaM C-lobe anchor residue of PKD2L1, and we predicted PKD2L1 channel activity regulation model by CaM by comparing the channel activity at various calcium concentrations. This study contributes to understanding the physiological role of PKD2L1 in diseases such as polycystic kidney disease, intestinal failure and congenital kyphosis.
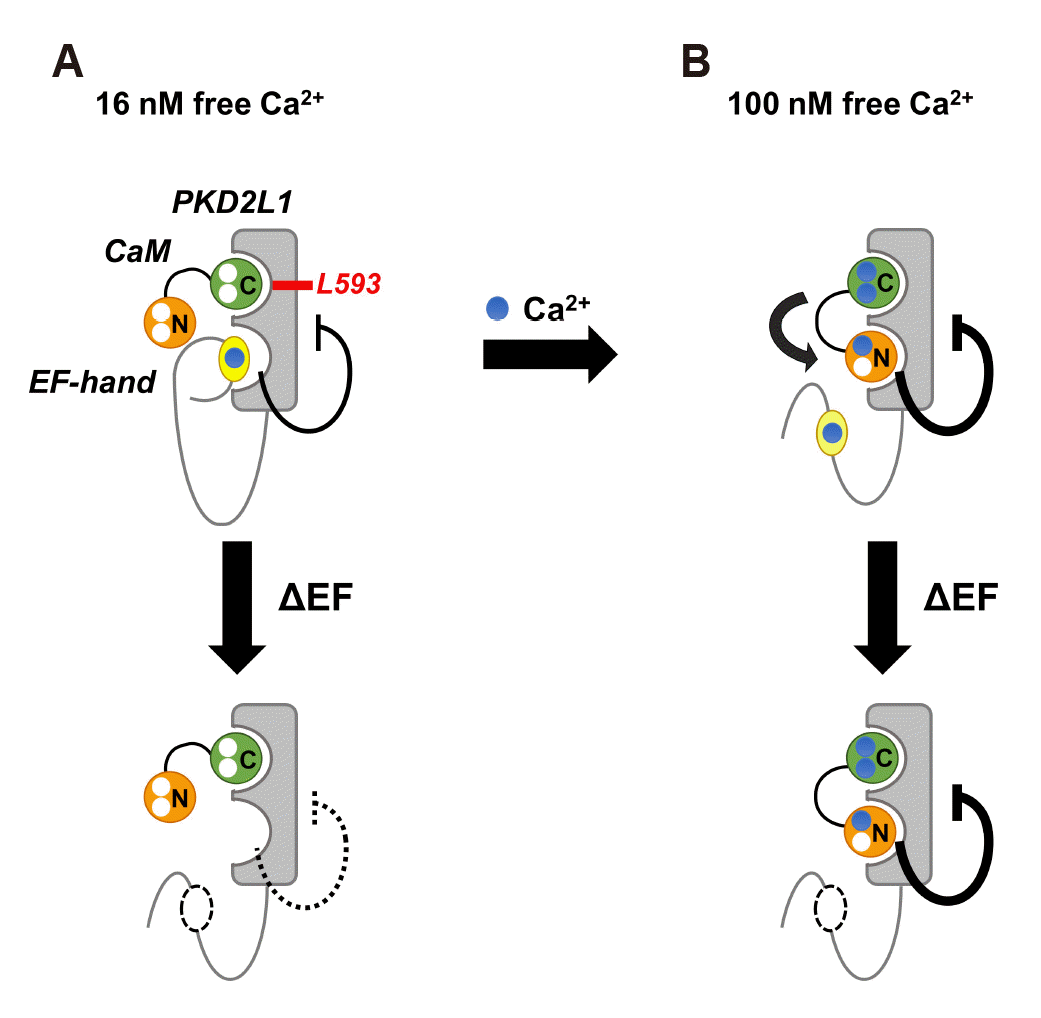
Fig. 7
Model of the mechanism by which calmodulin (CaM) regulates polycystic kidney disease 2-like-1 (PKD2L1).
(A) In 16 nM free calcium (blue), EF-hand (yellow) constantly binds to channel and shows weak inhibition. CaM C-lobe (green) is bound to PKD2L1 through Leu-593, a CaM C-lobe anchor residue (upper panel). At this time, if EF-hand deletion occurs, PKD2L1 inhibition by EF-hand is weakened (lower panel). (B) When calcium increases to 100 nM, CaM N-lobe (orange) binds to PKD2L1 instead of EF-hand, resulting in strong PKD2L1 inhibition (upper panel). Even with EF-hand deletion, CaM N-lobe still binds to PKD2L1 and strongly inhibits its activity (lower panel). The thick line indicates strong action.
![]()
 XML Download
XML Download