

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Promyelocytic leukemia (PML) gene was originally identified in patients suffering from acute promyelocytic leukemia (APL), associated with a chromosomal translocation of the PML gene on chromosomes 15 and the retinoic acid receptor alpha (RARα) gene on chromosome 17, generating the fusion protein PML-RARα [1,2]. A single PML gene, which includes nine exons, generates several PML isoforms through alternative splicing of exons 4–9 in the C-terminal region, which are seven major isoforms, including six nuclear PML isoforms designated PML-I to PML-VI and one cytoplasmic isoform, PML-VIIb [3]. All PML isoforms share the same N-terminal region containing the tripartite motif (TRIM) family consisting of cysteine-rich zinc-binding domains, a RING finger, two B-boxes (B1 and B2), and α-helical coiled-coil domain, named RBCC motif, which is involved in PML-nuclear bodies (PML-NBs) formation [3]. Although the PML isoforms may have related functions due to their common functional RBCC/TRIM domain, each PML isoform can interact with specific partners and possess distinct functions mediated by its specific C-terminal sequence [3].
PML is a major component of PML-NBs, which are nuclear matrix-associated domain and spheres of 0.1–1.0 μm in diameter found in most cell lines and many tissues [4]. In normal circumstances and particularly, in response to cellular stress, PML-NBs undergo significant changes in the number, size, and position [5]. PML recruits proteins to PML-NBs and regulates various cellular functions such as the induction of apoptosis, cellular senescence, inhibition of proliferation, maintenance of genomic stability, and antiviral response to mediate protein-protein interaction, resulting in PML-NBs mediate tumor suppression pathways [5,6]. Especially, p53-mediated cellular processes such as apoptosis, cell cycle arrest, DNA repair, and senescence occur in PML-NBs where PML enhances p53 protein stability by sequestering Mdm2 [6].
Signal transducer and activator of transcription-3 (STAT-3) was first identified as a DNA-binding factor from interleukin (IL)-6-stimulated hepatocytes that selectively binds to the IL-6-responsive element in the promoter of acute-phase genes [7]. STAT-3 in the cytoplasm of unstimulated cells is activated by binding several cytokines and growth factors, including epidermal growth factor, platelet-derived growth factor, and IL-6 to their receptors or non-receptor tyrosine kinases such as JAK and Src [8]. Once activated STAT-3 is phosphorylated on the tyrosine 705 residue, phosphorylated STAT-3 forms homodimers or heterodimers and translocates to the nucleus, consequently, STAT-3 dimers bind to specific promoter elements of target genes and regulate gene expression in the nucleus [8]. Activated STAT-3 induces the transcription of a broad panel of genes encoding regulator of cellular proliferation (such as cyclin D1 and MYC) and survival (such as BCL-xL and survivin) as well as angiogenesis-promoting (such as VEGF) and immunosuppressive growth factors and cytokines (such as IL-6) [9]. Although IL-6 is the most well-known classical activator of STAT-3, other IL-6 family members such as IL-11, IL-31, and oncostatin M (OSM) are involved in biological activities including inflammation, remodeling of the extracellular matrix, and modulation of cell growth and differentiation [10,11]. While physiological STAT-3 activation is transient, rapidly returning to the basal state, STAT-3 becomes inappropriately and persistently activated in a wide variety of hematopoietic and solid malignancies, including melanoma, multiple myeloma, breast, prostate, ovarian, and colon cancer [12]. Among the IL-6 family of cytokines, OSM was originally described as a novel and biological cancer therapy because of its ability to inhibit the growth of melanoma cells as well as breast cells, lung cells, and glioma cells. Despite the many studies implicating OSM as a suppressor of normal cell and select tumor cell proliferation, OSM has also been implicated, paradoxically, in cancer progression [13]. Also, it was well known that STAT-3 is activated in a high percentage of glioblastoma and recurrent glioblastoma exhibit increased phosphorylated STAT-3 levels when compared with primary glioblastoma [14].
It was well known that both PML and p53 are involved in apoptosis, senescence, differentiation, and cell cycle arrest by interacting with each other [15]. Previous studies showed that PML and p53 are involved in various cellular processes, PML inhibits STAT-3 activity, and STAT-3 and p53 interfere with each other. However, the interactions of PML, p53, and STAT-3 have not well known. In this study, the effect of the PML isoforms on OSM-induced STAT-3 transcriptional activity and how p53 was involved in these processes were examined.
Go to : 
METHODS
Cells
NIH3T3 mouse embryonic fibroblast cells, HEK293T human kidney epithelial cells, U87-MG and U251-MG human glioblastoma cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA). All cell lines were maintained in Dulbecco’s modified eagle’s medium (DMEM; Welgene, Gyeongsan, Korea) containing 10% fetal bovine serum (FBS; Gibco, Grand Island, NY, USA), 100 units/ml penicillin, and 10 μg/ml streptomycin at 37˚C in a humidified 5% CO2 incubator.
Reagents and antibodies
Recombinant murine OSM (mOSM) and human OSM (hOSM) were purchased from R&D Systems (Minneapolis, MN, USA). Antibody against p-STAT-3 Y705 was obtained from Cell Signaling Technology (Danvers, MA, USA) and antibodies against PML (H-238), p53 (DO-1), STAT-3 (F-2) were purchased from Santa Cruz Biotechnology (Dallas, TX, USA). Anti-α-tubulin antibody was obtained from Sigma-Aldrich (St. Louis, MO, USA).
Plasmids
For overexpression of PML proteins, pCMV-Tag2B, pCMV-Tag2B-PML-I, pCMV-Tag2B-PML-III, pCMV-Tag2B-PML-IV were used. PML-IV deletion constructs (pCMV-Tag2B-PML-IV ∆571-633, pCMV-Tag2B-PML-IV ∆571-620, and pCMV-Tag2B-PML-IV ∆621-633) were generated. To construct plasmid pCMV-Tag2B-PML-IV ∆571-633, pCMV-Tag2B-PML-IV ∆571-620, and pCMV-Tag2B-PML-IV ∆621-633, pCMV-Tag2B digested with BamHI and HindIII was ligated to the PCR fragment that contains human PML-IV ∆571-633 (which does not contain exons 8a and 8b) cDNA, human PML-IV ∆571-620 (which does not contain exons 8a) cDNA, and human PML-IV ∆621-633 (which does not contain exons 8b) cDNA each. For overexpression of p53 proteins, pCMV-Neo-Bam, pCMV-Neo-Bam-p53 wt, pCMV-Neo-Bam-p53 R273H were purchased from Addgene (#16440, #16434, and #16439). The STAT-3-Luc reporter plasmid containing four copies of the STAT-binding site was purchased from Addgene (#8688).
Transient transfection and reporter assays
Cells were seed at a concentration of 1 × 105 cells (NIH3T3), 2 × 105 cells (HEK293T, U87-MG, and U251-MG) in 6-well plates the day before transfection. Cells were transfected using Lipofectamine LTX with PLUS (Invitrogen, Waltham, MA, USA) according to the manufacturer’s instructions. 1 μg of the indicated plasmid was transiently co-transfected with both 0.4 μg of STAT-3-Luc reporter plasmid and 0.1 μg of pCMV-β-galactosidase plasmid. Cells were allowed to recover for 24 h before treatment with 10 ng/ml of OSM for 24 h, and then luciferase activity was measured. The luciferase activity of each sample was normalized to β-galactosidase activity to yield relative luciferase activity (RLA).
Western blot analysis
Cells were lysed in RIPA buffer containing Xpert Protease Inhibitor cocktail (GenDEPOT, Katy, TX, USA). Lysates were centrifuged 12,000 rpm at 4˚C for 30 min and then the supernatants were collected and used. Proteins were separated by 8% SDS-polyacrylamide electrophoresis and transferred to PVDF membrane (Bio-rad, Hercules, CA, USA). The membranes were blocked by 5% skim milk in Tris-buffered saline containing 0.1% Tween-20 (TBS-T) buffer for 1 h at room temperature and probed with primary antibodies (1:1,000) overnight at 4˚C. Membranes were washed in TBS-T buffer and incubated for 1 h at room temperature with HRP-conjugated specific secondary antibodies. After membranes were washed in TBS-T buffer, blots were developed using the ECL system (Amersham, Buckinghamshire, UK) and detected by ImageQuant LAS 4000 (GE Healthcare, Chicago, IL, USA).
Statistical analysis
All experiments were performed at least three times and the data was expressed as the mean ± standard deviation (SD). All statistical analysis was analyzed with GraphPad Prism 5 software (GraphPad Prism Software, Inc., San Diego, CA, USA). Student’s t-test was performed for comparisons between sample groups, and statistically significant differences were defined as p-value < 0.05.
Go to :
RESULTS
Differential effect of PML on OSM-induced STAT-3 transcriptional activity
In this study, two non-cancerous cell lines (NIH3T3 and HEK293T) and two glioblastoma cell lines (U87-MG and U251-MG) were utilized for comparing the non-cancer versus cancer cell lines and detecting STAT-3 transcriptional activity. To examine whether PML isoforms affect STAT-3 transcriptional activity induced by OSM, cells were transiently transfected with three different PML isoforms (PML-I, PML-III, or PML-IV) and STAT-3-Luc reporter construct, treated with or without OSM (10 ng/ml) for 24 h, and analyzed by reporter assay. Overexpression of PML isoforms negatively regulated OSM-mediated STAT-3 transcriptional activity in NIH3T3 (Fig. 1A) and U87-MG cells (Fig. 1C). On the contrary, OSM-induced STAT-3 activation was significantly increased in HEK293T (Fig. 1B) and U251-MG cells (Fig. 1D). Through immunoblotting analyses, we found that phosphorylation of STAT-3 is influenced by OSM treatment, not PML overexpression. Also, we confirmed the efficiency of the overexpression of PML isoforms (Fig. 1, right panels). These results indicate that the OSM-induced STAT-3 activation in the presence of PML is cell type-specific, and is not influenced by the type of PML isoform present.
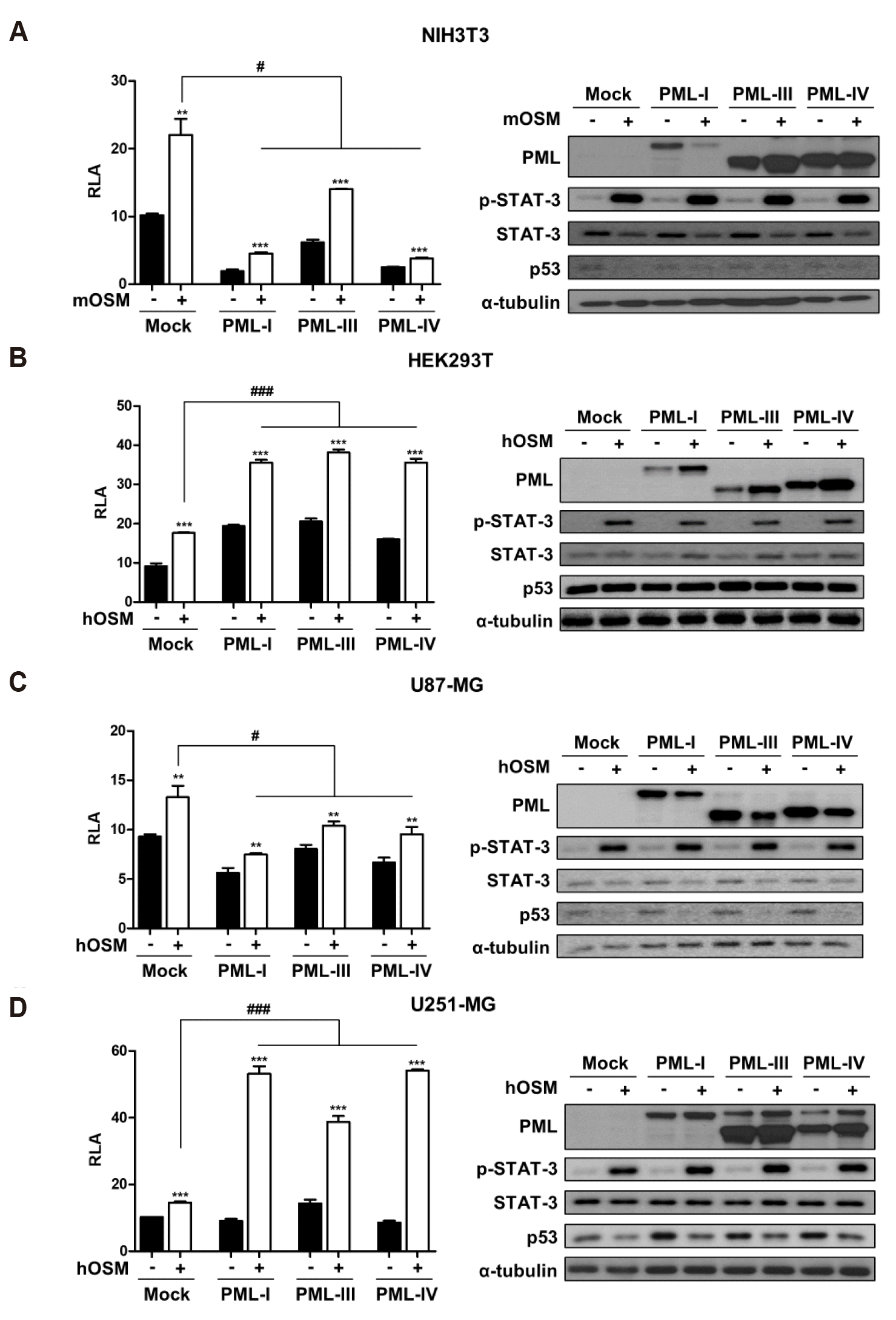 | Fig. 1PML affects differently OSM-induced STAT-3 transcriptional activity in immortalized cells and glioma cells.(A) NIH3T3, (B) HEK293T, (C) U87-MG, and (D) U251-MG cells were co-transfected with the STAT-3-Luc reporter and either empty vector or PML isoform expression vectors. At 24 h after transfection, cells were either untreated or treated with OSM (10 ng/ml) for 24 h and then assayed for luciferase activity. The pCMV-β-galactosidase vector was included to normalize transfection efficiency. Data are presented as fold increase in relative luciferase activity (RLA) compared with RLA in the absence of OSM. PML protein expression and OSM-induced STAT-3 phosphorylation were verified for each assay by immunoblotting. The results are representative of three independent experiments. mOSM, murine OSM; hOSM, human OSM. **p < 0.01, ***p < 0.001 vs. untreated control; #p < 0.05, ###p < 0.001 between cells were transfected with PML expression vectors and cells were transfected with mock vector in the presence of OSM.
|
Detection of endogenously expressed PML and wild-type and mutant p53
Since STAT-3 activity did not differ between PML isoforms and decreased or increased in a cell line-specific manner, the factor determining this cell line-specificity was investigated. It is well known that PML functions as a tumor suppressor along with p53. However, it is reported that PML can interact with mutant p53 and mediate cellular responses in a manner that is opposite to that of wild-type p53 [16]. To test the status of endogenous p53 and PML expression in each cell line, NIH3T3, HEK293T, U87-MG, and U251-MG cells were treated with or without OSM (10 ng/ml) for 24 h. According to the TP53 web site (http://p53.fr), NIH3T3 and U87-MG cells have wild-type p53, but U251-MG cells express p53 that is mutated at residue 273 (Arginine [R] to Histidine [H]) and HEK293T cells express inactive p53 because of SV-40 large T-antigen, and this information was tabulated in Fig. 2A. It is known that p53 has a very short protein half-life, and its protein levels are low in normal cells and tissues under non-stressed conditions. Conversely, mutant p53 proteins are typically stable and accumulate to very high levels in tumors [17]. Immunoblotting analysis revealed that NIH3T3 and U87-MG cells expressed wild-type p53, which was rarely detectable by immunoblotting, but HEK293T and U251-MG cells expressed the inactivated and mutant p53, which was strongly detected by anti-p53 antibody (Fig. 2B). When cells were treated with OSM for 24 h, p53 and STAT-3 expression were not changed by OSM treatment (Fig. 2B). However, interestingly, cells bearing mutant p53 showed strongly increased phosphorylation of STAT-3, compared to cells bearing wild-type p53 (Fig. 2B, compare lanes 4 and 8 with lanes 2 and 6). Since PML proteins exist in several isoforms in cells, multiple bands were detected on immunoblotting. Additionally, PML protein expression was increased in response to OSM.
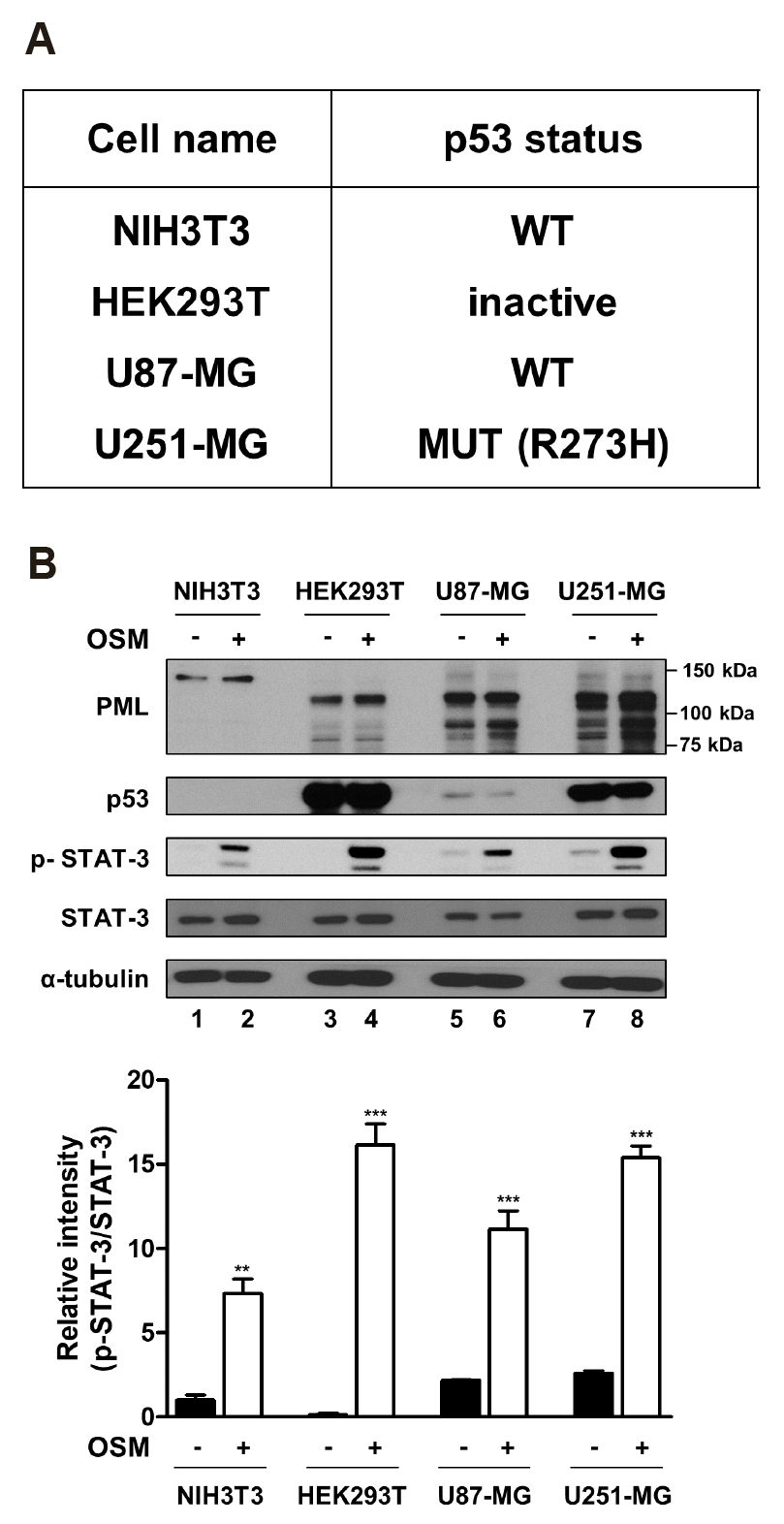 | Fig. 2The status of p53 and differential protein expression level.(A) The table indicates the p53 status of cell lines used in this study. p53 status was determined through the TP53 web site (http://p53.fr). (B, upper) NIH3T3, HEK293T, U87-MG, and U251-MG cells were either untreated or treated with OSM (10 ng/ml) for 24 h, and the lysed in RIPA buffer. Whole cell lysates were analyzed by immunoblotting using anti-PML, anti-p53, anti-p-STAT-3, anti-STAT-3, anti-α-tubulin antibodies. (B, lower) The graph represents the normalized intensities of p-STAT-3 against those of STAT-3 determined from three independent experiments. WT, wild-type. **p < 0.01, ***p < 0.001 vs. untreated control.
|
Opposite effects of PML-IV on OSM-mediated STAT-3 activity, depending on the p53 status
To understand the role of p53 on STAT-3 transcriptional activity in the presence of PML-IV, NIH3T3 and U87-MG cells were transiently transfected with either PML-IV or R273H-p53 or both, together with STAT-3-Luc construct (Fig. 3A, B). Similarly, HEK293T and U251-MG cells were transiently transfected with the STAT-3-Luc plasmid and with either PML-IV or p53-WT or both (Fig. 3C, D). When cells were overexpressed by PML-IV, OSM-induced STAT-3 activity corresponded with previous results (Fig. 1). In NIH3T3 and U87-MG cells, co-expression of PML-IV and R273H-p53 showed increased OSM-induced STAT-3 transcriptional activity, compared to the overexpression of PML-IV (Fig. 3A, B). Overexpression of R273H-p53 increased OSM-induced STAT-3 activity in NIH3T3 cells, whereas OSM-induced STAT-3 activity was not affected by the expression of R273H-p53 in U87-MG cells (Fig. 3A, B). As shown in Fig. 3C, D, co-expression of PML-IV and WT-p53 reduced OSM-induced STAT-3 transcriptional activity, compared to that on overexpression of PML-IV in HEK293T and U251-MG cells. OSM-induced STAT-3 activity was not affected by the expression of WT-p53 in HEK293T cells, whereas overexpression of WT-p53 decreased OSM-induced STAT-3 activity in U251-MG cells (Fig. 3C, D).
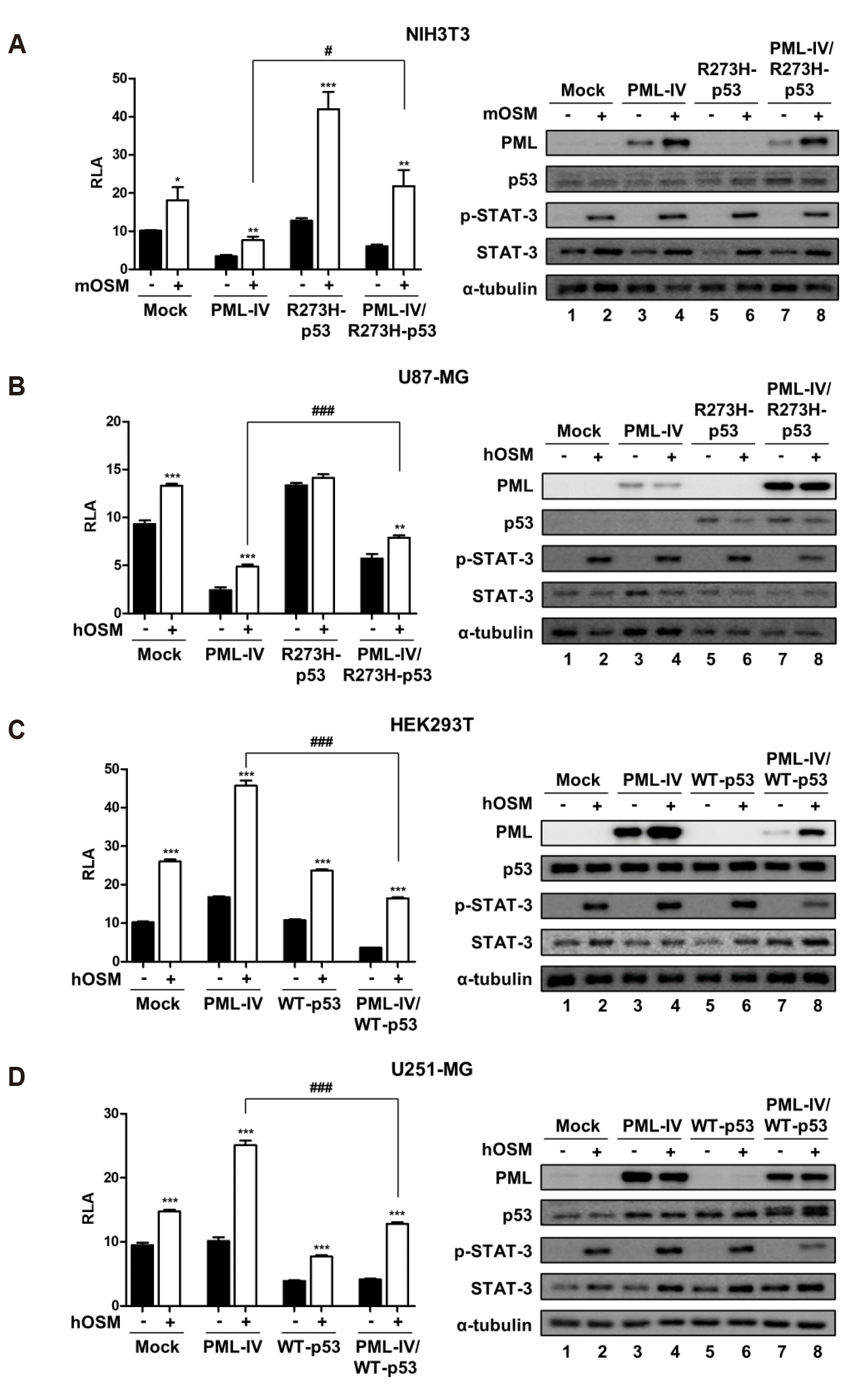 | Fig. 3The opposite effects of PML-IV on OSM-induced STAT-3 activity in p53-status dependent manner.(A) NIH3T3 and (B) U87-MG cells were transfected with either PML-IV or R273H-p53 or combination PML-IV and R273H-p53. (C) HEK293T and (D) U251-MG cells were transfected with either PML-IV or WT-p53 or combination PML-IV and WT-p53. At 24 h after transfection, cells were either untreated or treated with OSM (10 ng/ml) for 24 h and then assayed for luciferase activity. Data are presented as fold increase in relative luciferase activity (RLA) compared with RLA in the absence of OSM. PML and p53 protein expression and OSM-induced STAT-3 phosphorylation were verified for each assay by immunoblotting. The results are representative of three independent experiments. mOSM, murine OSM; hOSM, human OSM; WT, wild-type. *p < 0.05, **p < 0.01, ***p < 0.001 vs. untreated control; #p < 0.05, ###p < 0.001 between cells were transfected with PML-IV and cells were transfected with either combination PML-IV and R273H-p53 mutant or combination PML-IV and WT-p53 in the presence of OSM.
|
The effects of PML-IV and its deletion constructs on OSM-induced STAT-3 activity
A previous study showed that wild-type p53 binds to the C-terminal portion of PML-IV (amino acids 361-633) [18]. Therefore, we tested whether specific sequences of PML-IV affect STAT-3 transcriptional activity. For this purpose, three PML-IV constructs with deletions of specific exons were generated (Fig. 4A). Cells were transiently transfected with empty vector, PML-IV, PML-IV ∆571-633 (which does not contain exons 8a and 8b), PML-IV ∆571-620 (which does not contain exon 8a), PML-IV ∆621-633 (which does not contain exon 8b), and STAT-3-Luc construct. Next, cells were treated without or with OSM (10 ng/ml) for 24 h and analyzed by reporter assays. Consistent with the previous observation, overexpression of PML-IV resulted in decreased STAT-3 transcriptional activity in cells bearing wild-type p53 (NIH3T3 and U87-MG cells), and increased STAT-3 transcriptional activity in cells bearing mutant p53 (HEK293T and U251-MG cells) (Fig. 4B–E). As shown in Fig. 4B, overexpression of both PML-IV and three PML-IV deletion constructs reduced OSM-induced STAT-3 activity in NIH3T3 cells, especially PML-IV and PML-IV ∆621-633. Also, in U87-MG cells bearing wild-type p53, OSM-induced STAT-3 activity was significantly decreased when the constructs with PML-IV and PML-IV ∆621-633 mutant containing exon 8a were transfected (Fig. 4C). In contrast, in cells expressing mutant p53, both PML-IV and three PML-IV deletion constructs increased OSM-mediated STAT-3 transcriptional activity, compared to the control (Fig. 4D, E). These results indicate that in the presence of wild-type p53, both PML-IV and PML-IV deletion mutant bearing exon 8a regulate STAT-3 transcriptional activity in a negative manner. However, in the presence of mutant p53, STAT-3 transcriptional activity was increased by PML-IV and its deletion constructs, suggesting that increased STAT-3 activity in the presence of PML-IV was mediated by the common site of PML-I, PML-III, and PML-IV isoforms that include up to 7a.
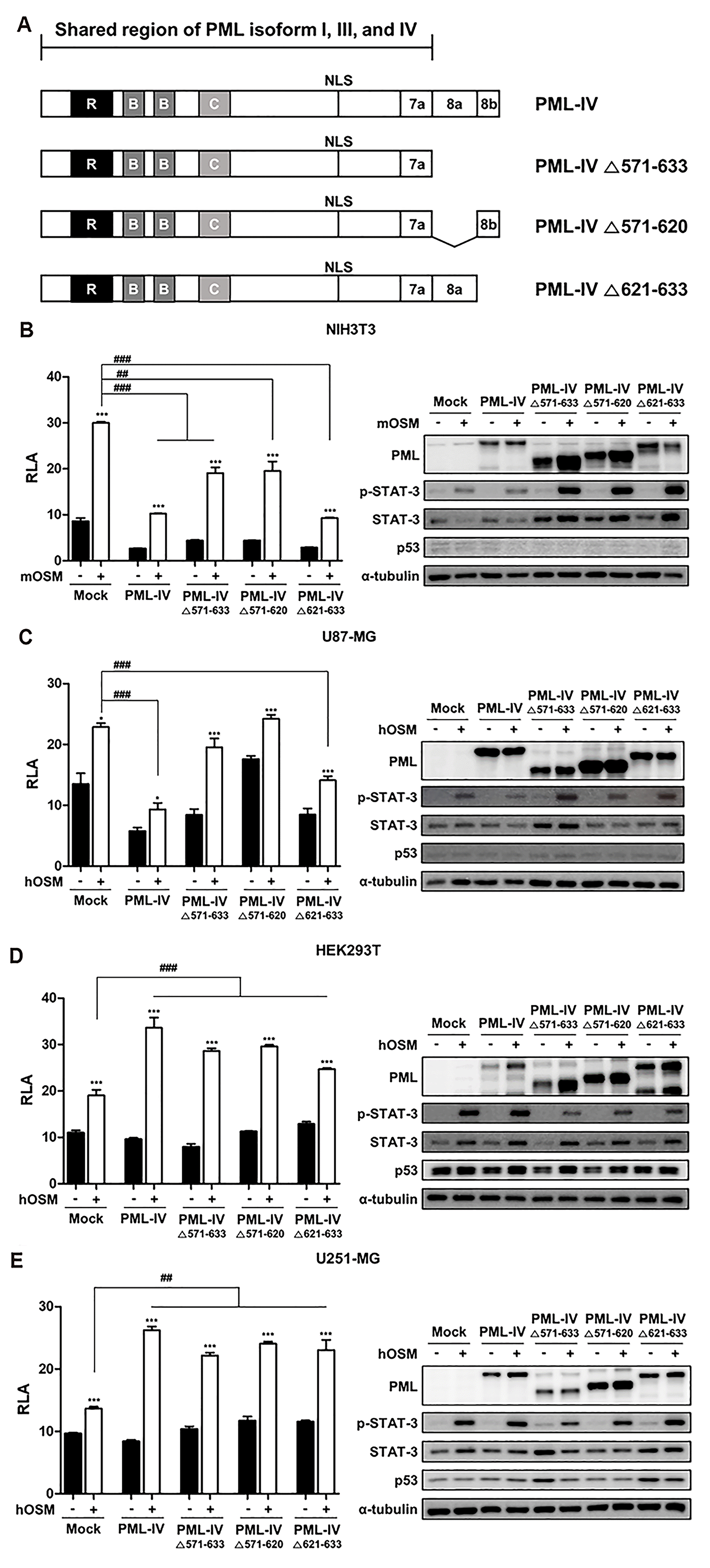 | Fig. 4The effects of PML-IV and its deletion constructs on OSM-mediated STAT-3 activity.(A) Depiction of the PML-IV deletion constructs generated in this study. (B) NIH3T3, (C) U87-MG, (D) HEK293T, (E) U251-MG cells were co-transfected with the STAT-3-Luc reporter and empty vector, PML-IV, and PML-IV deletion plasmids. At 24 h after transfection, cells were either untreated or treated with OSM (10 ng/ml) for 24 h and then assayed for luciferase activity. The pCMV-β-galactosidase vector was included to normalize transfection efficiency. Data are presented as fold increase in relative luciferase activity (RLA) compared with RLA in the absence of OSM. PML protein expression and OSM-induced STAT-3 phosphorylation were verified for each assay by immunoblotting. The results are representative of three independent experiments. mOSM, murine OSM; hOSM, human OSM. *p < 0.05, ***p < 0.001 vs. untreated control; ##p < 0.01. ###p < 0.001 between cells were transfected with either PML-IV or PML-IV deletion vectors and cells were transfected with mock vector in the presence of OSM.
|
Go to :
DISCUSSION
The results of the current study indicate that overexpression of the PML isoforms PML-I, PML-III, and PML-IV, exerted opposite effects on OSM-induced STAT-3 activity in cells expressing wild-type and mutant p53. The inhibitory effects of the PML isoforms were confirmed by a decrease in STAT-3 transcription activity in NIH3T3 and U87-MG cells, whereas overexpression of PML isoforms increased OSM-induced STAT-3 transcriptional activity in HEK293T and U251-MG cells (Fig. 1). However, it seems that PML overexpression itself regulates STAT-3 activity without OSM treatment. The previous study has been reported that PML binds to STAT-3 and inhibits STAT-3 DNA binding activity through B-box and C-terminal domains both basal status and IL-6-stimulated status [19]. Along with these results, we also observed that PML can regulate STAT-3 activity both in the presence and absence of OSM. Moreover, it has been reported that PML inhibits IL-6-induced STAT-3 activity by interfering with STAT-3 and HDAC-3 interaction [20]. In accordance with previous studies, we found that STAT-3 activity is regulated by PML in presence and absence of OSM treatment. But, the mechanism of regulation of STAT-3 activity by the PML isoforms was not suggested in the previous papers. Several studies are being done on PML isoforms, and as a result, different isoform functions are also being reported. For example, PML-I, PML-III, and PML-IV isoforms represses IFN-γ-induced STAT-1α transcriptional activity [21]. Ten-eleven translocation dioxygenase 2 (TET2) could bind all PML isoforms except PML-V, notably, PML-IV showed the most ability to bind to TET2 [22]. Also, PML-I and PML-IV bind to Smad2/3, but in the presence of TGF-β only PML-IV binds to phospho-Smad2/3, results in PML-IV overexpression promotes epithelial to mesenchymal (EMT) in MCF-7 tumor cells [23]. Till date, studies pertaining to PML isoform functions were focused largely on PML-IV. PML-IV is known to be involved in cellular processes by interacting with various proteins such as Myc, telomerase reverse transcriptase (TERT), EGFR, ARF, and SUMO-1, but the well-known protein for interacting with PML-IV is p53 [18,24-28]. These results show that PML isoforms can perform different functions, but, in this study, we did not observe the difference between PML isoforms, PML-I, PML-III, and PML-IV, at least on OSM-induced STAT-3 activity.
Wild-type p53 has a short protein half-life and its protein levels are generally low. On the contrary, the half-life of the mutant p53 is prolonged, conferring stability to the molecule, allowing it to accumulate in tissues [17]. As shown in Fig. 2, the cells harboring mutant p53 showed increased p53 protein expression, compared to wild-type p53 in immunoblotting. Moreover, increased phosphorylation of STAT-3 was observed in cells bearing mutant p53, compared to cells bearing wild-type p53. Furthermore, previous studies have reported that expression of wild-type p53 significantly reduced tyrosine phosphorylation of STAT-3 and inhibited STAT-3 DNA binding activity [29]. The STAT-3 protein binds to the p53 promoter and contributes to repression of the p53 promoter transcriptional activity. Therefore, inhibition of STAT-3 activity in tumor cells harboring wild-type p53 may provide a therapeutic advantage [30]. Several researchers have reported that the phosphorylation of STAT-3 correlated with p53 mutation and infiltrating immune cells in human pancreatic tumors [31]. Further, mutant p53 has been reported to activate STAT-3 phosphorylation in human colorectal cancer cells, resulting in EMT and cell migration [32]. Similar to previous studies, we observed that wild-type p53 inhibited phosphorylation of STAT-3 in response to OSM, and mutant p53 promoted the phosphorylation of STAT-3. Therefore, mutant p53 probably assumes a new role of regulating OSM-induced STAT-3 phosphorylation, independent of PML isoform overexpression.
Furthermore, it has been reported that PML is not only an upstream regulator of p53 but also acts as a general p53 effector, mediating p53 tumor suppressor functions [33]. On the contrary, PML enhances the transcriptional activity of mutant p53 and is required for the proliferation of cancer cells bearing mutant p53 [16]. When wild-type p53 cells were co-transfected with both PML-IV and R273H-p53 mutant, OSM-mediated STAT-3 transcriptional activity was significantly enhanced, compared to that of cells which were transfected with PML-IV expression vector alone (Fig. 3A, B); However, when cells bearing mutant p53 were co-transfected with both PML-IV and wild-type p53, OSM-induced STAT-3 transcriptional activity was significantly decreased, compared to that of transfected cells with PML-IV alone (Fig. 3C, D). These results strongly suggest that wild-type or mutant p53 and PML-IV act together to regulate STAT-3 activity, resulting in increased or decreased STAT-3 transcriptional activity, depending on the p53 status. Additionally, these results provide evidence that the interaction of PML and mutant p53 may contribute to aberrant activation of STAT-3 in cancer cells. Interestingly, it was found that endogenously expressed mutant p53 is able to increase phosphorylation of STAT-3 (Fig. 2, lanes 4 and 8); however, overexpression of either mutant p53 or wild-type p53 does not change phosphorylation of STAT-3 in cells bearing either wild-type p53 or mutant p53 (Fig. 3, lane 6). Overexpression of both PML-IV and mutant p53 did not change STAT-3 phosphorylation in cells bearing wild-type p53 (Fig. 3A, B, lane 8). In contrast, overexpression of both PML-IV and wild-type p53 decreased phosphorylation of STAT-3 in cells bearing mutant p53 (Fig. 3C, D, lane 8). These results indicate that the relationship of STAT-3 phosphorylation by OSM and STAT-3 transcriptional activity by overexpression of PML and wild-type p53 is significant only in cells bearing mutant p53 endogenously (Fig. 3C, D).
In conclusion, PML regulates STAT-3 transcriptional activity, which is dependent on the p53 status rather than PML isoforms, at least in response to OSM. Further research needs to be conducted on how PML isoform affects and works for other STATs. Overexpression of PML-I, PML-III, and PML-IV decreased OSM-induced STAT-3 transcriptional activity in cells bearing wild-type p53, whereas, overexpression of PML-I, PML-III, and PML-IV increased OSM-mediated STAT-3 transcriptional activity in cells harboring mutant p53. PML acts as a tumor suppressor and interferes with the transcriptional activity of STAT-3 in the presence of wild-type p53. On the other hand, when mutant p53 is present, PML does not act as a tumor suppressor, thereby increasing the transcriptional activity of STAT-3. Consequently, STAT-3 suppression activity of PML protein has the reliance on the status of p53.
Go to :
 XML Download
XML Download