

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Lung cancer is a major cause of morbidity and mortality worldwide and is one of the leading causes of cancer-related deaths [1]. Non-small-cell lung cancer (NSCLC) is a type of lung cancer that includes squamous carcinoma, adenocarcinoma, and large-cell carcinoma. Compared with small-cell carcinoma, NSCLC cells grow more slowly and metastasize relatively late. NSCLC accounts for approximately 80% to 85% of the total number of lung cancer cases and is currently treated mainly using chemotherapy [2]. However, severe toxicity greatly limits chemotherapy. Therefore, there is an urgent need to develop a safe and effective adjuvant therapy for NSCLC.
Sesamin is a lipid-soluble lignin that was first found in sesame seeds and has since been identified in 30 different species of plants. Detailed studies have shown that sesamin has anticancer effects, which are mainly attributable to its anti-proliferative, anti-metastatic, anti-inflammatory, pro-apoptotic, and pro-autophagocytic properties [3]. In recent years, several studies have reported the anticancer effects of sesamin on a variety of cancers, such as breast, prostate, cervical, and lung cancer [4-6]. Sesamin was reported to induce apoptosis in human lung cancer cells through inhibition of the mitochondrial protease Lon and activation of the DNA-damage checkpoints [7]. Recent research suggested that sesamin blocks the phosphoinositide 3-kinase (PI3K)/Akt signaling pathway in lung cancer by downregulation of the expression of cyclooxygenase 2 (COX2), leading to cell cycle arrest and apoptosis [8]. A recent study reported that sesamin suppressed NSCLC cell proliferation and induced G1 arrest of the cell cycle and apoptosis via the Akt/p53 pathway [9]. Although previous studies have confirmed that sesamin can inhibit proliferation and promote the apoptosis of NSCLC, the mechanism is still not completely clear. Our has previously reported that sesamin could induce autophagic death in cervical cancer cells [6]; therefore, we hypothesized that mitochondrial autophagy may be involved in the inhibitory effect of sesamin on lung cancer.
Oxidative phosphorylation of mitochondria is an efficient process that is accompanied by excessive reactive oxygen species (ROS) production [10]. Excessive ROS in the body damages mitochondria and promotes the release of apoptosis-promoting factors, eventually leading to cell death [11]. To maintain mitochondrial homeostasis, damaged mitochondria need to be removed in time to promote mitochondrial renewal. Mitochondrial renewal in cells is mainly through the selective autophagy pathway. In this process, the dysfunction and aging mitochondria are selectively degraded, which is called mitophagy [12]. The early manifestation of mitophagy is mitochondrial depolarization, after which mitochondria are specifically enveloped in autophagosomes, which then fuse with lysosomes to complete the degradation and renewal of mitochondria [13]. Mitochondrial membrane potential collapse is a prerequisite and early event for the PINK1/Parkin pathway activation and mitophagy. When the mitochondrial membrane potential disintegrates, PINK1 stably accumulates on the mitochondrial outer membrane, activating the transfer of Parkin from the cytoplasm to the mitochondria, and eventually inducing mitophagy [14].
In this study, we used the A549 cell line to investigate the inhibitory effect of sesamin on NSCLC. We provide evidence to support the therapeutic role of sesamin in lung cancer through induction of oxidative stress-mediated apoptosis and mitophagy, further confirming its anticancer properties.
METHODS
Cell culture
The A549 lung adenocarcinoma cell line was obtained from the Cell Resource Center of the Shanghai Institute of Life Sciences, Chinese Academy of Sciences. Cells were cultured in complete growth medium consisting of Dulbecco’s modified Eagle’s medium (DMEM; Gibco, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS; Gibco), 100 g/ml streptomycin, and 100 U/ml penicillin at 37°C in a humidified 5% CO2 and 95% air atmosphere.
Cell viability assay
Cell viability was determined using the Cell Counting Kit-8 (CCK-8; Dojindo, Kumamoto, Japan) as described by the manufacturer. Briefly, 2 × 103 cells per well were plated in 96-well culture plates and incubated in 100 µl of complete growth medium. The synergistic effects of sesamin on human lung cancer A549 cell line viability were assessed by treating cells with 10, 20, 40, 60, 80, and 100 µM sesamin (purity > 98%; Chengdu Must Bio, Chengdu, China) dissolved in chloroform (Amresco, Solon, OH, USA) for 12, 24, 36, and 48 h; cells treated with vehicle (0.1% chloroform) were used as controls. After treatment, 10 µl CCK-8 solution was added to each well and plates were incubated for an additional 4 h at 37°C. The percentage of viable cells was determined by measuring optical density in triplicate wells at 450 nm using a microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Scratch-wound test
The effects of sesamin on the migration of A549 cells was evaluated using the scratch-wound test. For this test, 2 × 105 cells per well were seeded in 6-well plates, and then incubated overnight to allow cells to cover the entire plate. Thereafter, a “wound” was made by scratching the monolayer, and plates were washed three times with phosphate-buffered saline (PBS). Cells were then treated with 20 or 40 µM sesamin, or vehicle (0.1% chloroform), in DMEM for 48 h. Images were acquired at 0 and 48 h using an inverted microscope at 200× magnification. The wound-closure rate was calculated according to the formula, ([0 h scratch width – 48 h scratch width] / [0 h scratch width]) × 100%.
Transmission electron microscopy
A549 cells were seeded in 6-well plates and cultured in DMEM containing 40 µM sesamin or 0.1% chloroform (vehicle control) at 37°C and 5% CO2 for 48 h. Cells were collected by centrifugation at 300× g for 10 min at room temperature and fixed in 2.5% glutaraldehyde for 1 h. Next, cells were fixed in a 1% osmium tetroxide solution for 1 h, dehydrated with a graded series of ethanol washes (30%, 60%, 70%, 90%, and 100%), which infiltrated the cells, and then embedded in LR White resin. Ultrathin sections (50–75 nm), sliced with an LKB ultramicrotome, were stained with 2% aqueous uranyl acetate and 2% aqueous lead citrate, and transmission electron microscopy (TEM) images were obtained at 5,000× magnification.
Terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay
Apoptotic DNA fragmentation was detected by fluorescence microscopy using a DeadEnd Fluorometric TUNEL System kit (Promega, Wisconsin, WI, USA). A549 cells were seeded at 1 × 105 cells/well in 6-well plates and grown in a humidified atmosphere of 5% CO2 at 37°C. After treating cells with 40 µM sesamin and/or the ROS scavenger N-acetyl-L-cysteine (NAC, 5 µM; Sigma-Aldrich) for 48 h, TUNEL assays were performed according to the manufacturer’s instructions. Samples were immediately analyzed under a fluorescence microscope using standard filter sets for fluorescein (green) at 520 ± 20 nm and propidium iodide (PI; red) at > 620 nm.
Cell Mitochondria Isolation
Cell Mitochondria Isolation and cytoplasmic protein extraction were measured using a Cell Mitochondria Isolation Kit (Beyotime, Shanghai, China) as described by the manufacturer. Briefly, cells were collected by centrifugation at 100 g after digestion with trypsin cell digestive solution, and precipitated with cold PBS and then centrifuged at 600 g and 4°C for 5 min. After discarding the supernatant, add 1 ml of mitochondrial separation reagent to 2 × 107 cells, gently suspend the cells and place them in ice bath for 10 min. Next, transfer the cell suspension to a glass homogenizer of appropriate size for about 30 times. The cell homogenate was centrifuged at 600 g and 4°C for 10 min. The supernatant was then transferred to another centrifuge tube and centrifuged at 11,000 g for 10 min at 4°C. The precipitate is the isolated cell mitochondria. Carefully collect the supernatant and centrifugate it at 4°C for 10 min and he supernatant is cytoplasmic protein (mitochondria removed).
Western blotting
Treated cells were washed twice with ice-cold PBS (pH 7.4) and lysed in RIPA buffer (Solarbio, Beijing, China) supplemented with phenylmethylsulfonyl fluoride (PMSF; Solarbio) and a phosphatase inhibitor (Roche, Basel, Switzerland). Lysates were incubated for 5 min on ice and then microcentrifuge at 14,000 rpm for 10 min at 4°C. Protein concentration in whole-cell lysates was quantified using a BCA kit (Mbchem, Shanghai, China), resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE), and then transferred to polyvinylidene difluoride (PVDF) membranes (Millipore, Bedford, MA, USA). Non-specific binding was blocked by incubating membranes with TBST (Tris-buffered saline with 0.1% Tween-20) containing 5% non-fat milk for 2 h at room temperature. Thereafter, membranes were incubated overnight at 4°C with primary antibodies (diluted in TBST) against cleaved caspase-3, cleaved caspase-9, cytochrome c (1:1,000; all from Cell Signaling, Beverly, MA, USA); and Parkin (1:1,000), PINK1 (1:2,000), Bax (1:1,000), B-cell lymphoma-2 (Bcl-2) (1:1,000), β-Tubulin (1:2,000), VDAC1 (1:2,000) and GAPDH (1:5,000, internal control; all from Santa Cruz Biotechnology, Santa Cruz, CA, USA). Membranes were then washed and incubated with appropriate horseradish peroxidase-conjugated secondary antibodies (Santa Cruz Biotechnology) for 1 h at room temperature. Finally, protein signals were detected using enhanced chemiluminescence (ECM) (Western Blotting Kit; Boster, Wuhan, China).
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential (Δψm) was measured using a JC-1 assay kit (Beyotime) as described by the manufacturer. Briefly, A549 cells were seeded at a density of 5 × 105 per well in 6-well plates, incubated for 12 h, and then treated with 40 µM sesamin and/or 5 mM NAC for 48 h. Following removal of the culture medium, 1 ml of cell culture medium and 1 ml of JC-1 dye buffer were added, fully mixed, and incubated at 37°C for 20 min. The supernatant was removed and cells were washed twice with JC-1 dye buffer (1×). Washed cells were resuspended in culture medium (2 ml) and observed under a fluorescence microscope.
Intracellular ROS measurement
A549 cells were plated in 6-well plates and incubated for 12 h to allow cells to reach the exponential growth phase. Cells were then treated with 40 µM sesamin (or 0.1% chloroform) and/or 5 mM NAC, at 37°C and 5% CO2 for 48 h, after which they were harvested by centrifugation for 5 min at 37°C and resuspended in DCFH-DA (10 µM) for 30 min at 37°C in the dark. The intracellular H2O2 content was determined by flow cytometry (BD Biosystems, Franklin lake, NJ, USA) and analyzed using Cell Quest software (BD Biosystems).
Flow cytometry
Apoptosis was examined by staining cells with an Annexin V Apoptosis detection kit I (BD Biosciences, San Jose, CA, USA) as described by the manufacturer. Briefly, cells were washed twice with cold PBS and resuspended in 1× Binding Buffer at a concentration of 1 × 106 cells/ml. Subsequently, 100 µl cell solution (1 × 105 cells) was mixed with 5 µl Annexin V and 5 µl PI and incubated for 15 min at room temperature in the dark. Finally, 400 µl of 1× Binding Buffer was added to each tube, and apoptosis was analyzed within 1 h using a FACScan flow cytometer (BD Biosystems).
Cell cycle was determined with a cell cycle detection kit (Beyotime) according to the manufacturer’s protocol. In brief, A549 cells were washed twice with cold PBS and fixed with 70% ethanol at 4°C for 2 h, Following PBS washing for 3 times and 30 min of PI staining at 37°C. Finally, cell cycle was analyzed within 1 h using a FACScan flow cytometer (BD Biosystems).
Statistical analysis
The data presented are expressed as means ± standard deviation (SD) of three independent experiments performed in triplicate. The significance of differences between two means was determined by Student’s t-test using SPSS 19.0 software (IBM Co., Armonk, NY, USA). p-values < 0.05 were considered statistically significant.
RESULTS
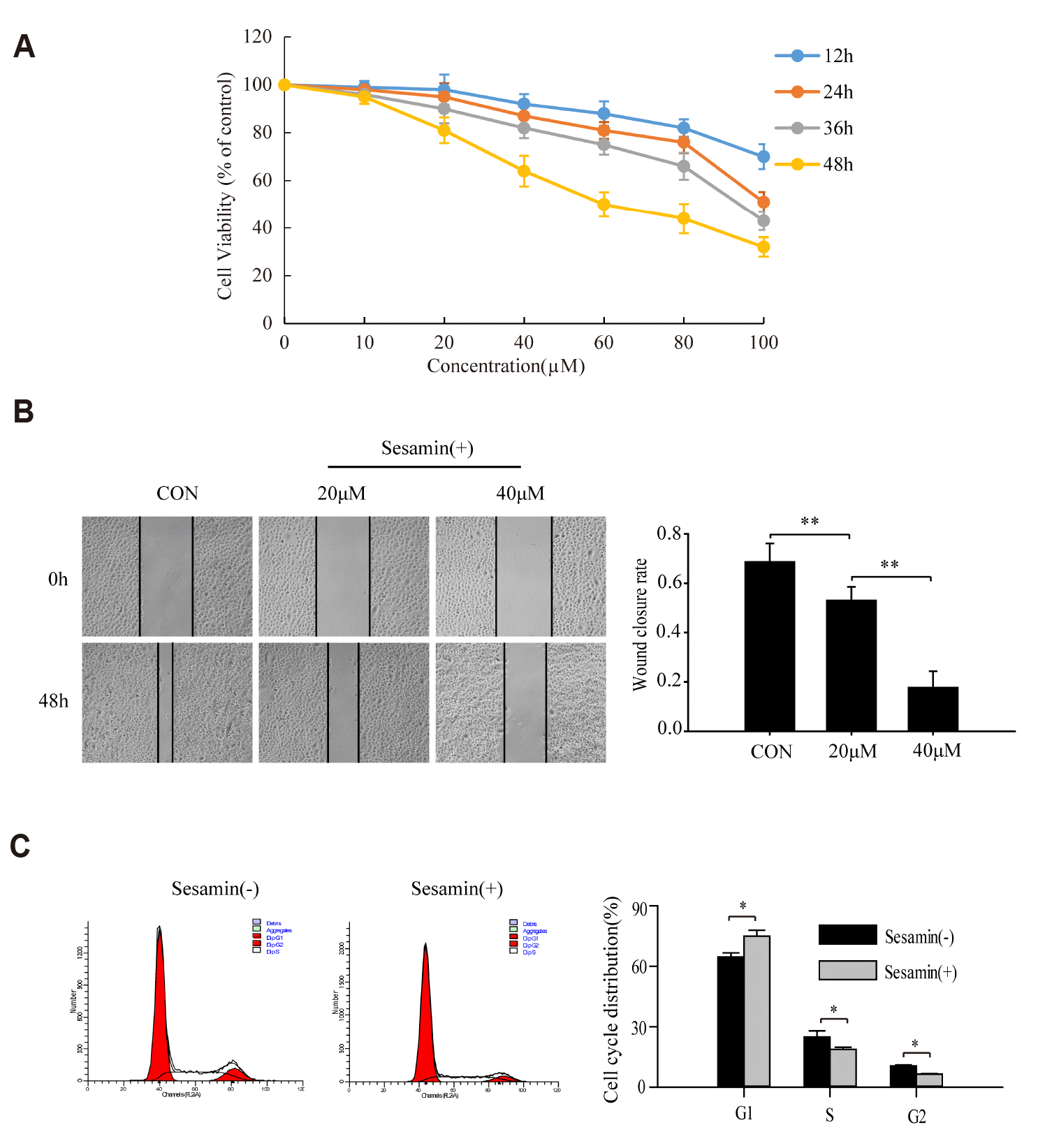
Sesamin inhibits the proliferation and migration of human lung cancer cells
A549 cells were treated with 10, 20, 40, 60, 80, and 100 µM sesamin, or 0.1% chloroform (vehicle control), for 12, 24, 36, and 48 h, and cell viability was assessed using CCK-8 assays. These analyses showed that sesamin exerted a time- and concentration-dependent depressant effect on the proliferation of A549 cells (p < 0.05) (Fig. 1A). The effect of sesamin on the migration ability of A549 cells was assessed by treating cells with 20 or 40 µM sesamin (or 0.1% chloroform) for 24 and 48 h, and then performing scratch-wound assays. These experiments showed that the migratory ability of sesamin-treated A549 cells, measured as the degree of wound closure, was lower than that of the control group (Fig. 1B). A549 cells were treated with 40 µM sesamin (or 0.1% chloroform) for 48 h, and then detected the cell cycle by flow cytometry, the results showed that the cells in G2 and S phase decreased and blocked in G0/G1 phase after sesamin treatment (Fig. 1C), further indicating the suppressive effect of sesamin on the proliferation and migration of A549 cells.
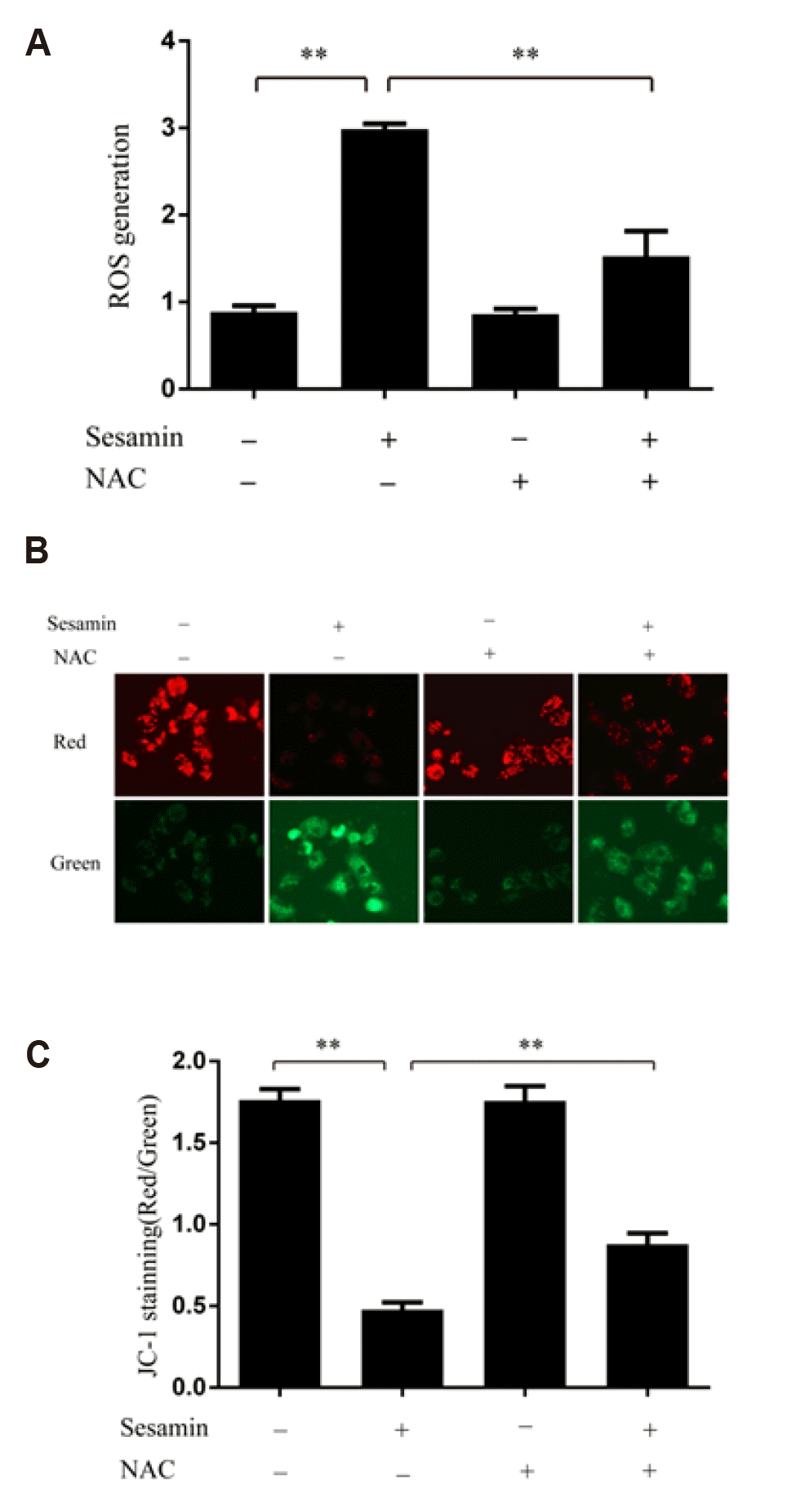
Sesamin triggers the collapse of mitochondrial membrane potential in A549 cells by increasing ROS levels
To establish whether sesamin stimulates intracellular oxidative stress, we examined ROS levels based on DCFH-DA fluorescence using flow cytometry. As shown in Fig. 2A, 40 µM sesamin significantly increased ROS generation in A549 cells. This increase was significantly attenuated by pretreatment of A549 cells with the antioxidant NAC (5 mM; p < 0.05). It is well known that ROS accumulation can cause mitochondrial damage. Accordingly, we measured changes in mitochondrial membrane potential using a JC-1 assay kit containing the cationic dye, which undergoes a readily detectable shift from red to green with decreases in membrane potential. As shown in Fig. 2B and C, sesamin treatment increased the green/red fluorescence ratio in A549 cells, an effect that was prominently reduced by 5 mM NAC, indicating that sesamin triggers the collapse of mitochondrial membrane potential through ROS accumulation.
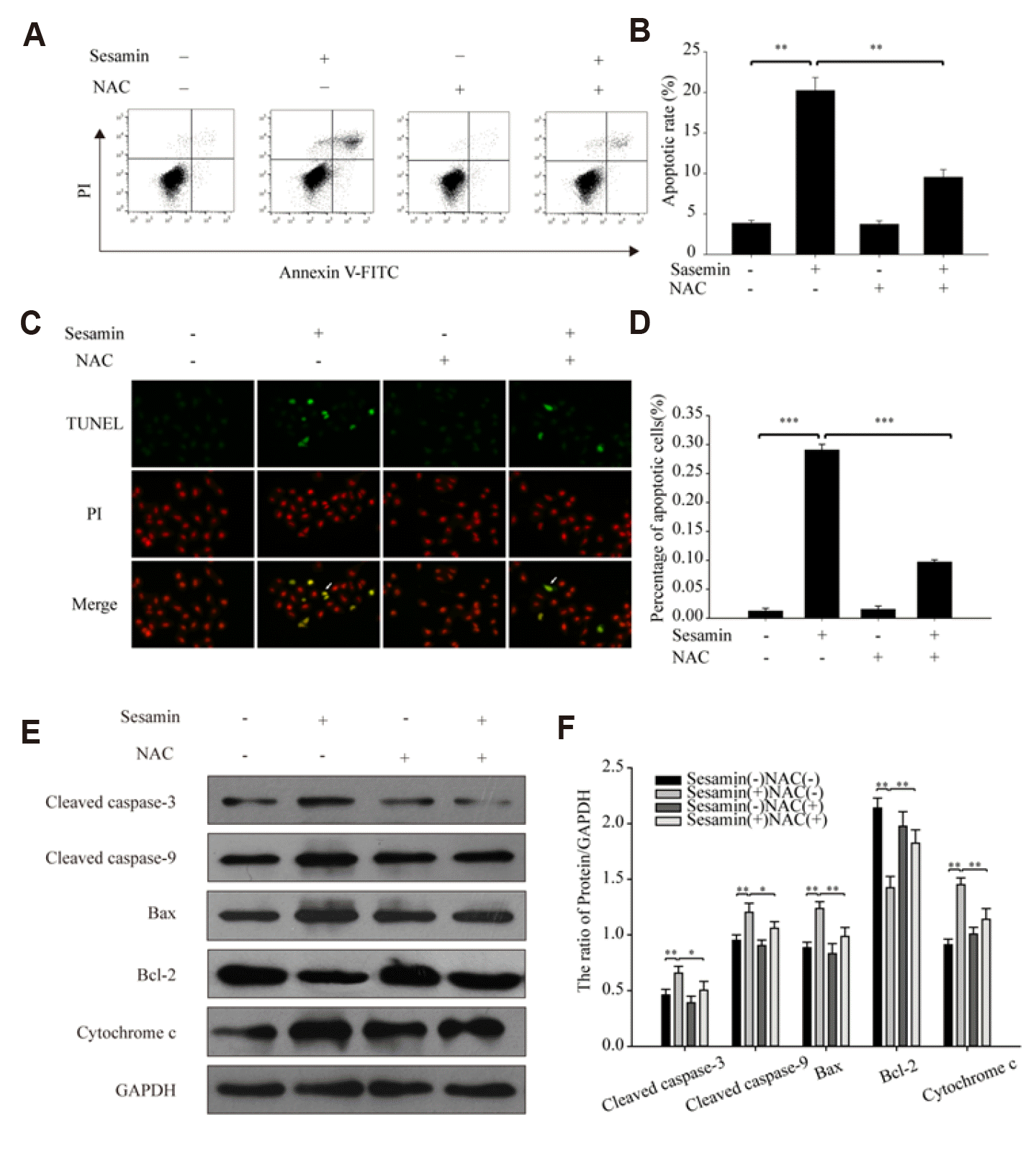
Sesamin acts through a ROS-dependent mitochondrial pathway to induce apoptosis of A549 cells
To investigate whether sesamin induces A549 cell apoptosis, we used flow cytometry and TUNEL staining. A549 cells were treated with 40 µM sesamin for 48 h, after which apoptosis was assessed by flow cytometric detection of TUNEL-stained cells. These experiments showed that sesamin promoted A549 cell apoptosis, and that 5 mM NAC alleviated this effect (Fig. 3A–D). Next, we assessed the expression of the mitochondrial apoptosis-related proteins, cleaved caspase-3, cleaved caspase-9, Bax, Bcl-2, and cytochrome c by Western blotting. This analysis showed that sesamin treatment increased the expression of cleaved caspase-3, cleaved caspase-9, cytochrome c and the ratio of Bax/Bcl-2 in A549 cells; notably, these increases were block by pretreatment with 5 mM NAC (Fig. 3E, F). Taken together, our data suggest that sesamin induces A549 cell apoptosis via a ROS-dependent mitochondrial pathway.
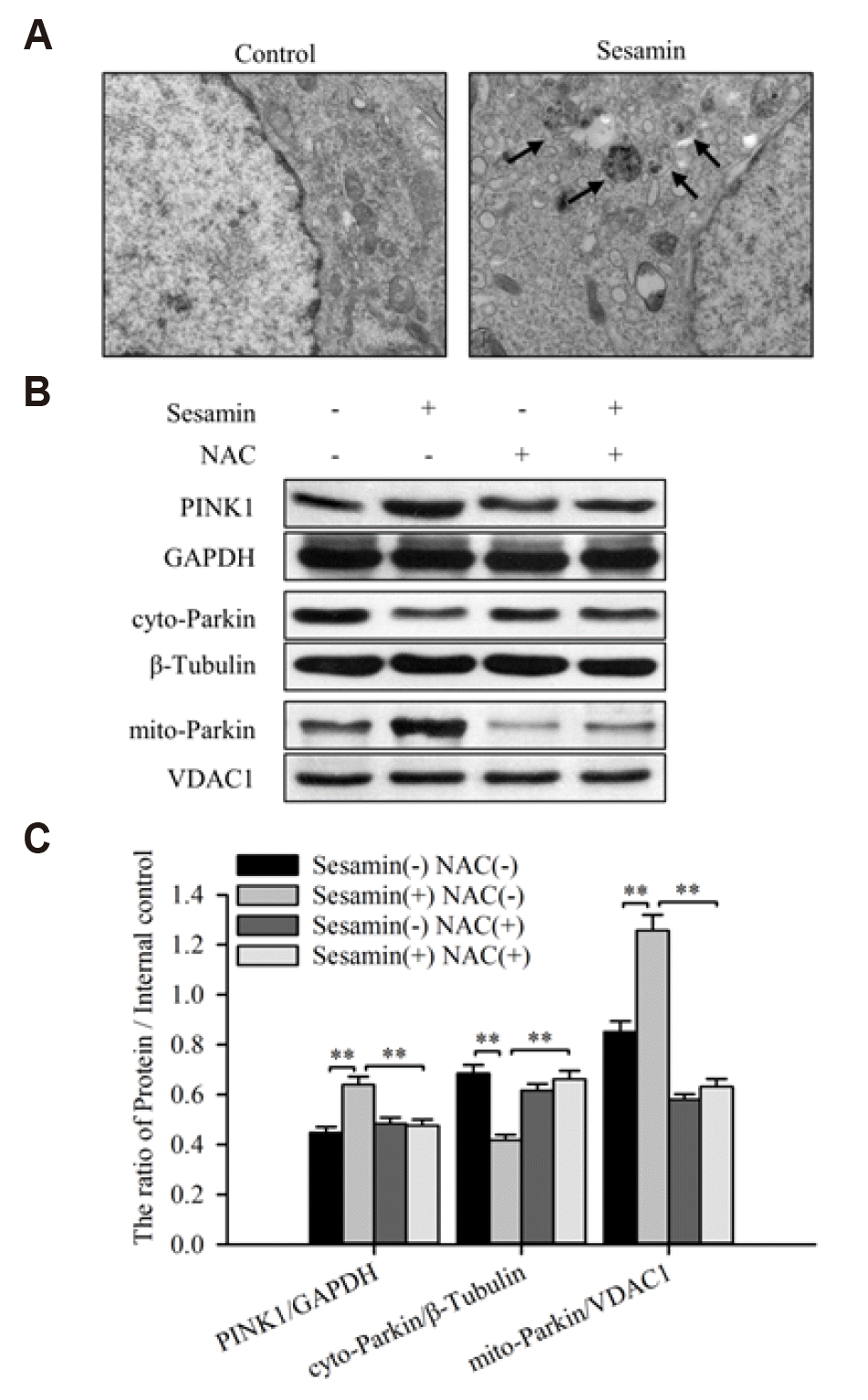
Sesamin triggers mitophagy in A549 cells through a ROS-mediated PINK1/Parkin pathway
Decreases in mitochondrial membrane potential may lead to mitophagy, a process that involves the PINK1/Parkin pathway [15,16]. To investigate whether sesamin triggers mitophagy, we used TEM to examine ultrastructural changes in A549 cells treated with 40 µM sesamin for 48 h. As shown in Fig. 4A, mitochondrial swelling and degeneration, and numerous autophagosomes containing mitochondria, were observed under the electron microscope, confirming that sesamin treatment induces mitophagy. To further investigate effect of sesamin on mitophagy, we next examined changes of PINK1 in whole cell and Parkin in mitochondria and cytoplasm.by Western blotting. As shown in Fig. 4B, C, sesamin increased the expression of PINK1 in whole cell and Parkin in mitochondria, but decreased the level of Parkin in cytoplasm, indicating the translocation of Parkin from cytoplasm to mitochondria. In addition, NAC blocked the up-regulation of PINK1 and the translocation of Parkin induced by sesamin, indicating a role for ROS in the induction of mitophagy by sesamin.
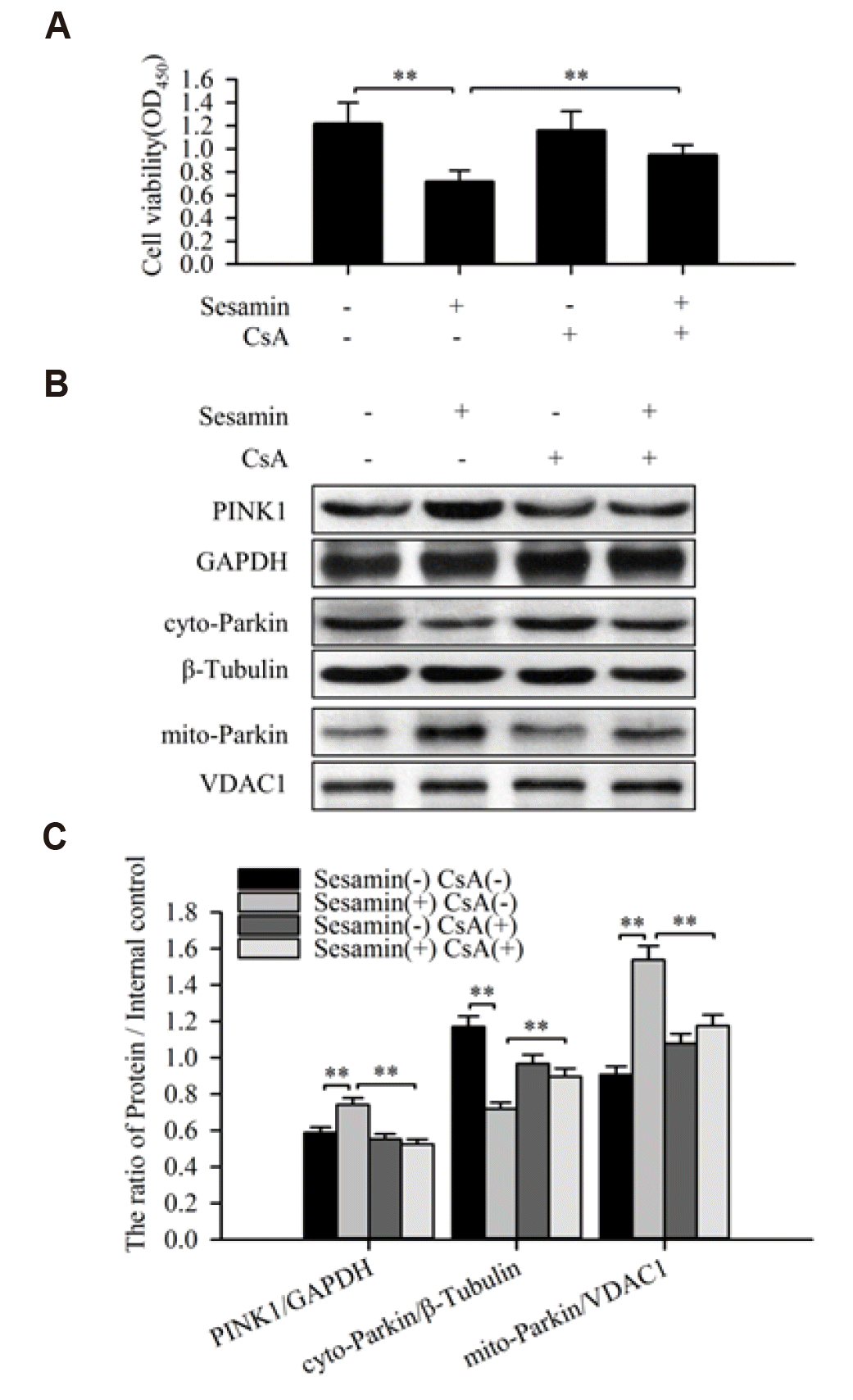
Inhibition of mitophagy weakens the inhibitory effect of sesamin on A549 cell viability
Cyclosporine A (CsA), a specific inhibitor of mitophagy, is capable of inhibiting mitochondrial depolarization and mitochondrial autophagosome formation by interfering with the interaction of cyclophilin D with the mitochondrial permeability transition pore [17]. To explore the role of mitophagy in sesamin-treated A549 cells, we blocked mitophagy with 5 µM CsA and detected the expression of PINK1 in whole cell and Parkin in mitochondria and cytoplasm by Western blotting. This analysis showed that CsA inhibited the sesamin-induced increases in PINK1 in whole cell and Parkin in mitochondria. In addition, compared with the sesamin treated group, the expression of Parkin in the cytoplasm (sesamin and CsA combined treatment group) increased, indicating Parkin translocating from cytoplasm to mitochondria decreased (Fig. 5B, C). Tests of cell proliferation ability using the CCK-8 assay, the result further showed that CsA (5 µM) reduced the inhibitory effect of sesamin on A549 cell proliferation (Fig. 5A).
DISCUSSION
Sesamin, a type of lipid-soluble lignin originally isolated from sesame seeds, has since been found in more than 30 different plants, including willow, pepper, and magnolia [18]. A number of studies have shown that sesamin possesses bioactive properties, including anti-inflammatory and anticancer activity [3]. In 2010, Wang and colleagues reported that obtusilactone A and (-)-sesamin induce apoptosis in human lung cancer cells by inhibiting the mitochondrial protease Lon and activating DNA damage checkpoints [7]. A more recent study showed that sesamin blocks the PI3K/pAKT signaling pathway in lung cancer by down-regulating the expression of COX2, leading to cell cycle arrest and induction of apoptosis [5]. In the current study, we found that sesamin inhibited the proliferation and migration of human lung cancer cell and induced oxidative stress in lung cancer cells, leading to a decline in mitochondrial membrane potential and subsequent induction of mitophagy and apoptosis.
ROS and antioxidants are key signaling molecules that participate in oxidative stress responses. Under specific pathological conditions, accumulation of excessive ROS can disrupt cellular homeostasis, leading to oxidative stress and mitochondrial dysfunction [19]. Mitochondria are the main sites of ROS production as well as targets of ROS. Mitochondria in cancer cells are both structurally and functionally distinct from those in normal cells, and actively participate in metabolic recombination, characterized by ROS overproduction, to promote cancer development [20]. Therefore, reducing intracellular ROS levels and weakening the survival advantage that they confer may represent an effective strategy for inhibiting tumor development. In addition, excessive levels of ROS cause serious damage to cellular components, including DNA, proteins, and mitochondrial lipid membranes [21]. Data presented here indicate that sesamin induces alterations in the antioxidant system that lead to elevated ROS levels in A549 cells.
Mitochondria are the main source of ROS, which in turn affect mitochondria by causing a breakdown in the mitochondrial membrane potential (Δψm). A decrease in Δψm is considered the earliest event in the apoptosis cascade. Following substantial changes in Δψm, members of the caspase protease family are activated, initiating an apoptotic cascade reaction and irreversible entry into the apoptotic pathway [22]. Measurement of Δψm in A549 cells using the JC-1 assay kit showed that sesamin disrupts Δψm, an effect that was attenuated by the antioxidant, NAC. These data indicate that sesamin triggers oxidative stress in A549 cells, resulting in a decrease in Δψm. In addition, measurements of apoptosis using flow cytometry and TUNEL assays showed that sesamin increased A549 cell apoptosis via a ROS-dependent mechanism.
Caspase-3 is present in cells as an inactive precursor under normal circumstances, but is cleaved under stress conditions to generate an active fragment that mediates apoptosis [23]. Cellular apoptosis can be divided into two main pathways: endogenous and exogenous. Exogenous apoptosis is mediated by cell death receptor-cleaved caspase-8, whereas endogenous apoptosis is mediated by cleaved caspase-9 [24]. Excessive accumulation of ROS promotes mitochondrial membrane permeability and collapse of the mitochondrial membrane potential. Cytochrome c, an important electron transport component of the respiratory chain, is subsequently released [25], and in turn activates caspase-9, which activates the caspase cascade, and ultimately caspase-3, to induce apoptosis [26]. Experiments from the present study showed that sesamin treatment promoted upregulation of cytochrome c and activation of caspase-3 and caspase-9. Notably, the increase in these proteins was blocked by pretreatment with NAC. Collectively, these findings indicate that sesamin induces apoptosis via the ROS-dependent intrinsic mitochondrial pathway.
Bcl-2 is a crucial cancer-related protein in apoptosis research. Bcl-2 inhibits cell death induced by a variety of cytotoxic factors. The earlier discovery that overexpression of Bcl-2 enhances the resistance of cells to most cytotoxins led to the hypothesis that various apoptotic signaling mechanisms have a common pathway or intersection point that is regulated by Bcl-2 [27]. Excessive Bcl-2 expression is associated with reduced generation of ROS. Previous studies support an indirect antioxidant effect of Bcl-2, such that Bcl-2 inhibits the production of superoxide anions rather than directly scavenging ROS. In addition, decreasing Bcl-2 levels alters the mitochondrial permeability transition pore structure and increases mitochondrial membrane permeability, causing breakdown of the mitochondrial membrane potential [28]. Bax, which also belongs to the Bcl-2 family, is the most important pro-apoptotic protein in the human body and exerts an inhibitory effect on Bcl-2. The Bax/Bcl-2 ratio is regarded as a key factor in determining whether cells enter the apoptotic pathway [29]. Our Western blot data confirmed that sesamin increases the Bax/Bcl-2 ratio. Taken together with previous experimental findings, our results suggest that sesamin induces A549 cell apoptosis via a ROS-dependent mitochondrial pathway.
Mitophagy is a type of autophagy process in which redundant and damaged mitochondria are selectively degraded, a concept first formally proposed by Lemasters [12] in 2005. During mitophagy, damaged mitochondria become isolated. Mitochondrial autophagocyte-related proteins are subsequently activated and accumulate on the surfaces of damaged mitochondria—specifically identifiable by the autophagosome membrane—and become encapsulated to form the mitochondrial autophagosome, which then fuses with the lysosome to form a mitochondrial autophagosome that mediates the degradation of damaged mitochondria [30]. Morphological changes in mitochondrial ultrastructure can be observed by TEM—the gold standard for the detection of mitochondrial autophagy. Using TEM to detect changes in mitochondrial ultrastructure in sesamin-treated A549 cells treatment, we found that a vacuolar double-layer membrane structure appeared around mitochondria, encapsulating them to form mitochondrial autophagosomes. These results indicate that sesamin induces mitophagy in A549 cells.
The PINK1/Parkin pathway has been identified as a paradigmatic mechanism for mammalian mitophagy. Under physiological conditions, dimeric PINK1 rapidly dissociates and is degraded in the intima after entering mitochondria, where it is maintained at a stable low level [31]. Studies have shown that, in the context of oxidative stress, PINK1 rapidly senses decreases in mitochondrial membrane potential characteristic of damaged mitochondria, and accumulates on the surface of the outer membrane of these mitochondria, where it induces accumulation of Parkin [32]. In the present study, TEM revealed the presence of many autophagosomes containing mitochondria in sesamin-treated A549 cells, suggesting that sesamin induces A549 cell mitophagy. We also found that sesamin increased the expression of PINK1 and induced the translocation of Parkin from cytoplasm to mitochondria. These effects were significantly attenuated by NAC, indicating that sesamin triggers mitophagy through a ROS-mediated PINK1/Parkin pathway in A549 cells.
Autophagic lysosomes selectively remove damaged, folded and redundant mitochondria, so as to maintain a balance between mitochondrial quality and quantity, and thus ensuring cell survival [33]. Thus, mitochondrial autophagy is generally thought to be a cell-protective mechanism. However, extensive autophagy also causes cytotoxic effects, leading to cell death [34]. Cyclosporin A is a specific inhibitor of mitochondrial permeability transport pathway, which plays a role by inhibiting the opening of mitochondrial permeability transport channels, and ultimately inhibits the cell apoptosis and necrosis dependent on mitochondrial permeability transport channels [35,36]. To explore the role of mitophagy in the effect of sesamin on A549 cells, we used CsA to block mitochondrial autophagy. We found that CsA diminished the cytotoxicity of sesamin toward A549 cells, indicating that sesamin induces A549 cell autophagic death.
 XML Download
XML Download