

 PDF
PDF Citation
Citation Print
Print


INTRODUCTION
Autophagy is an essential recycling process that delivers cytoplasmic material to the lysosome for degradation [1]. Growing evidence indicates that malfunction of autophagy often results in numerous disease symptoms due to an accumulation of toxic proteins in cells [2]. In the case of cancer, the dual role of autophagy in promoting cell survival or death has been widely studied [3]. Autophagy has been reported to play an essential death-promoting role in cells that are unable to undergo apoptosis [4]. Conversely, other laboratories have demonstrated a tumor-promoting role of autophagy [5]. Autophagy-mediated macromolecular degradation contributes to building block supply, which fuels the elevated metabolism of cancer cells [6]. Our previous studies have also shown the contradictory roles that autophagy plays in both promoting and suppressing carcinogenesis [7-9].
Paclitaxel, a microtubule-stabilizing drug, has been widely used as a chemotherapeutic drug against a variety of cancer types [10]. Paclitaxel has been known to induce cell death by inducing mitosis arrest. [11]. Despite its potent efficacy in anticancer therapy, the occurrence of multidrug resistance (MDR) limited paclitaxel efficacy in clinic [12]. Moreover, paclitaxel resistance mechanisms are complicated and have not been well characterized yet. Although overexpression of ATP-binding cassette (ABC) transporters, which effectively pump chemotherapy drugs out of the tumor cells [13], is an important mechanism inducing resistance to paclitaxel [14], it has also been known that many other factors may be responsible for the resistance to this drug. Several proposed mechanisms of action include MAPKs activation [15], changes in PI3K/mTOR signaling [16], cell cycle deregulation [17], and modulation of histone acetyltransferase activity [18].
In recent years, it has been reported that biphasic changes in autophagy contribute to resistance development in response to paclitaxel treatment [19]. We also previously reported that autophagy modulators was able to improve the therapeutic of paclitaxel [20]. However, little is known about the molecular mechanisms underlying autophagy induction in tumor cells during paclitaxel resistance development. In this study, to investigate the role of autophagy in multidrug resistant v-Ha-ras-transformed NIH 3T3 (Ras-NIH 3T3/Mdr) cells, we generated knockout (KO) Ras-NIH 3T3/Mdr cells that were deficient in ATG5 using CRISPR/Cas9 system [21,22]. ATG5 is an E3 ubiquitin ligase essential for autophagosome elongation [23,24]. This study demonstrates that paclitaxel suppresses the proliferation of ATG5 KO cells more markedly than their parental MDR cells, suggesting a pro-survival role of autophagy in MDR cells after the treatment of paclitaxel.
Go to : 
METHODS
Reagents and antibodies
The RNeasy Midi Kit was purchased from Qiagen (Valencia, CA, USA). SYBR Premix EX Taq II and WST-1 were acquired from Takara Korea Biomedical Inc. (Seoul, Korea). Fetal bovine serum (FBS), Dulbecco’s modified Eagle’s medium (DMEM), and lipofectamine 2000 were purchased from Thermo Fisher Scientific (Waltham, CA, USA). Anti-ATG5 antibody was acquired from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Anti-LC3 antibody, paclitaxel, hydroxychloroquine, and rapamycin were purchased from Sigma-Aldrich (St. Louis, MO, USA). SBI-0206965 was obtained from Cell Signaling Technology (Danvers, MA, USA).
Cell lines and culture conditions
The development of Ras-NIH 3T3/Mdr cells, which show high levels of P-glycoprotein (P-gp) compared with their parental counterparts (Ras-NIH 3T3 cells), has been previously described [25]. The Ras-NIH 3T3/Mdr cells were maintained at 37°C in DMEM supplemented with 10% FCS. The Ras-NIH 3T3/Mdr cells were passaged at least three times in paclitaxel-free culture medium before use in assays. Paclitaxel was made up in dimethyl sulfoxide (DMSO) as a stock solution and freshly diluted in culture medium before each experiment. The final concentration of DMSO in all the experiments never exceeds 0.1%.
Plasmid DNA and transient transfection
ATG5 CRISPR/Cas9 construct were obtained from ToolGen (Seoul, Korea). pEGFP-LC3 (Addgene #11546), pCI-neo-mAtg5 (Addgene #22956) and ptfLC3 (Addgene #21074) were obtained from Addgene (Cambridge, MA, USA). The cells were transiently transfected by Lipofectamine 2000 with an expression vector encoding pEGF-LC3 or pCI-neo-mAtg5. At 24 h post-transfection, cells were treated with paclitaxel.
Establishment of the ATG5 KO cell line
ATG5 KO cell lines were generated with ATG5 CRISPR/Cas9 construct as previously described [9], with target single guide (sg) RNA sequence: 5΄-AAGATGTGCTTCGAGATGTGTGG-3΄. The expanded single cell clones were used for assessment of ATG5 gene status. The following primer sets were used to confirm ATG5 KO: forward primer, 5΄-GCTTCGAGATGTGTGGTTTG-3΄ and reverse primer, 5΄-CAGTGGTGTGCCTTCATATT-3΄. The PCR products were verified by agarose gel electrophoresis (2.0% [w/v] agarose) followed by staining with ethidium bromide.
Quantitative reverse transcription PCR (RT-qPCR) analysis
The mRNA levels of four ABC transporters were measured by RT-qPCR. Briefly, cDNA weas used for qPCR containing primers specific for each ABC transporter. All primers were synthesized by Bioneer (Daejeon, Korea). The primer sequences used for the qPCR analysis are listed in Table 1. The qPCR was carried out with an Applied Biosystems 7300 Real-Time PCR System (Foster City, CA, USA). The qPCR data were evaluated by the 2−ΔΔCt method [26], normalized by the expression of β-actin.
Table 1
Primer sequence for real-time quantitative PCR analysis
![]()
Autophagy assay
Autophagy was detected by conversion of LC3-I to LC3-II using immunoblot analysis. Immunoblotting was performed with monoclonal LC3 antibody as previously described [27]. Immunoblot images were captured using the Bio-Rad ChemiDoc XRS+ system (Hercules, CA, USA), and image analysis was done using the Bio-Rad Image Lab software version 5.2.1 (Bio-Rad Laboratories).
Cell survival assay
WST-1 assay was carried out to measure the cytotoxicity of paclitaxel on ATG5 KO and their parental MDR cells. The cells were seeded in quadruplicate wells of 96-well plates and were then treated with paclitaxel for 2 or 3 days. A volume of 10 μl of WST-1 was added to each well and incubated for 4 more hours at 37°C. The absorbance at 450 nm was measured using a SpectraMax 190 microplate reader (Molecular Devices, Sunnyvale, CA, USA).
Cell cycle analysis by flow cytometry
Trypsinized cells were ethanol-fixed and then stained for total DNA with propidium iodide (PI) for 5 min. The DNA content was measured with the Gallios flow cytometer (Beckman Coulter, Inc., Brea, CA, USA). Data were acquired with the Kaluza analysis software (Beckman Coulter, Inc.).
Apoptotic assay by flow cytometry
Apoptotic assay was carried out using the FITC Annexin V Apoptosis Detection Kit (BD Biosciences Pharmingen, San Jose, CA, USA). Briefly, harvested cells (2 × 106) were resuspended the cells in binding buffer and incubated with PI and FITC-conjugated Annexin-V for 10 min in the dark. After adding the binding buffer, the samples were immediately analyzed by the Gallios flow cytometer with the Kaluza analysis software (Beckman Coulter, Inc.).
Go to :
RESULTS
Generation and validation of a Ras-NIH 3T3/Mdr ATG5 KO cell line
We established ATG5 KO in Ras-NIH 3T3/Mdr cell lines using CRISPR/Cas9 technology, as we previously reported [27]. We designed a sgRNA targeting exon 2 of the ATG5 gene (Fig. 1A). RT-PCR analysis revealed a loss of ATG5 mRNA in one clone (clone 3) from six initial clones (Fig. 1B). Autophagy deficiency in clone 3 was also confirmed by blockage of conversion of soluble LC3-I to lipid bound LC3-II induced by rapamycin, which is a powerful autophagy inducer (Fig. 1C). The lipidation of LC3-I into LC3-II is a well-recognized hallmark to evaluate the degree of autophagy activity [28]. We further verified autophagy deficiency in ATG5 KO cells by transfecting a tandem mRFP-GFP-LC3 reporter (Fig. 1D). Yellow fluorescence, which represented co-localization of RFP fluorescence with GFP, indicates an autophagosome, and a RFP-only signal is considered an autolysosome [29]. In parental MDR cells (ATG5 wild-type [WT]), rapamycin induced the formation of autophagosomes and autolysosomes. However, the numbers of GFP-LC3 and RFP-LC3 puncta were remarkably reduced in ATG5 KO cells as compared with WT cells under both control and rapamycin-treated conditions.
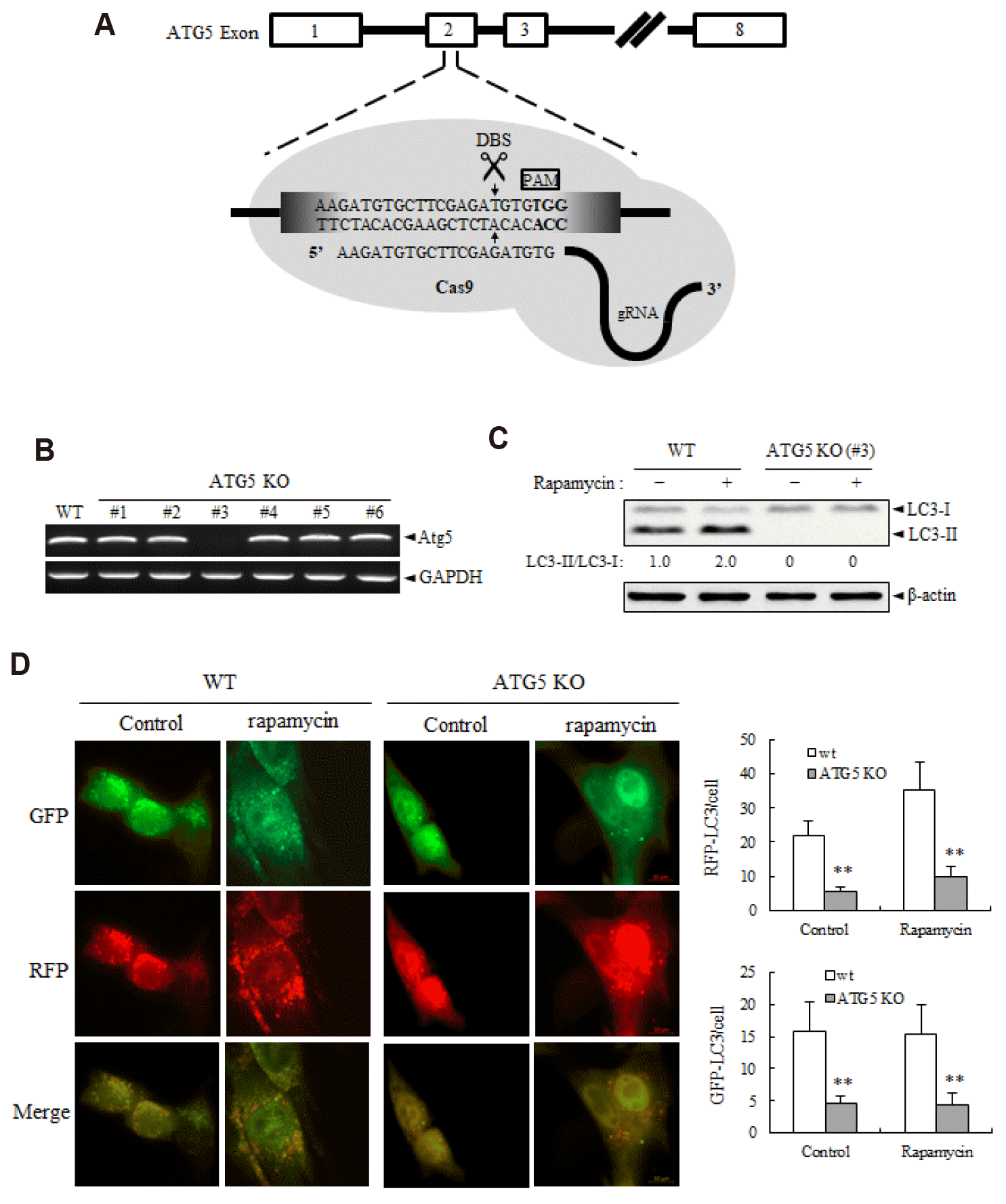 | Fig. 1Validation of the ATG5 knockout (KO) in Ras-NIH 3T3/Mdr cells.(A) The sgRNA target site is located at the exon 2 of ATG5 gene. The 20-nt guide sequence comprising the 5΄-end of the chimeric sgRNA is shown. (B) RT-PCR was performed to confirm KO of ATG5. PCR products were by agarose gel electrophoresis (2.0% [w/v] agarose). GAPDH was used as a internal control. (C) To confirm autophagy deficiency in the ATG5 KO cell line, cells were treated with rapamycin (250 nM). The conversion of soluble LC3-I to lipid bound LC3-II was identified by immunoblotting. β-Actin expression was used as a protein loading control. Results shown here are representative of at least three independent experiments. (D) Autophagic flux were measured using the ptfLC3 plasmid that simultaneously expresses RFP- and GFP-tagged LC3 protein. (D, left panel) Representative images showing GFP-LC3 and mRFP-LC3 puncta. Early autophagosomes show GFP-RFP colocalization whereas late, acidic autophagosomes lose GFP and appear red. (D, right panel) Quantitative analysis of GFP-LC3 puncta and RFP-LC3 puncta. Data are expressed as mean ± SD. DSB, double-strand break; WT, wild-type. **p < 0.01 as compared with ATG5 WT cells, as determined by Dunnett’s t-test.
|
ATG5 KO restores paclitaxel sensitivity in MDR cells
As expected, Ras-NIH 3T3/Mdr cells showed stronger resistance to paclitaxel than Ras-NIH 3T3 cells (Fig. 2A). Interestingly, Ras-NIH 3T3/Mdr cells with ATG5 KO was re-sensitized to paclitaxel, suggesting that functional autophagy may be required for paclitaxel resistance in MDR cells. The involvement of autophagy in paclitaxel resistance was further confirmed by the addition of early and late autophagy inhibitors (Fig. 2B). SBI-0206965 has been shown to effectively inhibit autophagy at early-stage when targeting autophagy kinase ULK1 [30], whereas hydroxychloroquine (HCQ) has been known to block late-stage autophagic flux [31]. The influence of two autophagy inhibitors treatment on cell growth compared to control was measured over a 72-h time course. We found that treatment with either SBI-0206965 or HCQ could significantly sensitize Ras-NIH 3T3/Mdr cells to paclitaxel at concentrations as low as 300 nM. An early autophagy inhibitor SBI-0206965 was more effective in inhibiting cell survival than a late autophagy inhibitor HCQ. The ABC family of transporters has been known to be involved in transport-based classical MDR [32]. Among 49 different types of transporters [32], P-glycoprotein (P-gp/ABCB1), MDR protein 1 (MRP1/ABCC1), breast cancer resistant protein (BCRP/ABCG2), and MDR 10 (ABCC10/MRP7) transporters were selected because they are known to frequently drive chemo-sensitive cancers to MDR [33,34]. Thus, to investigate the effect of ATG5 KO on the expression of these transporters, the mRNA expression levels of four ABC transporters were determined using RT-qPCR analysis (Fig. 2C). Ras-NIH 3T3 cells were used as paclitaxel-sensitive control cells. There was no correlation between resistance to paclitaxel and expression of MRP1, MRP7 and BCRP proteins, regardless of whether they were either ATG5 KO cells or their parental MDR cells. Meanwhile, the expression level of P-gp, which is greatly increased in cells with acquired MDR, decreased significantly after the ATG5 KO. Interestingly, we also observed that AGT5 KO cell line derived from a non-resistant parent cells (Ras-NIH 3T3 ATG5 KO/Mdr), which was further manipulated to become MDR, showed a strong expression of P-gp comparable to that in Ras-NIH 3T3/Mdr cells. These results imply that the autophagy deficiency might not be the direct cause for downregulation of P-gp, as observed in Ras-NIH 3T3/Mdr ATG5 KO cells.
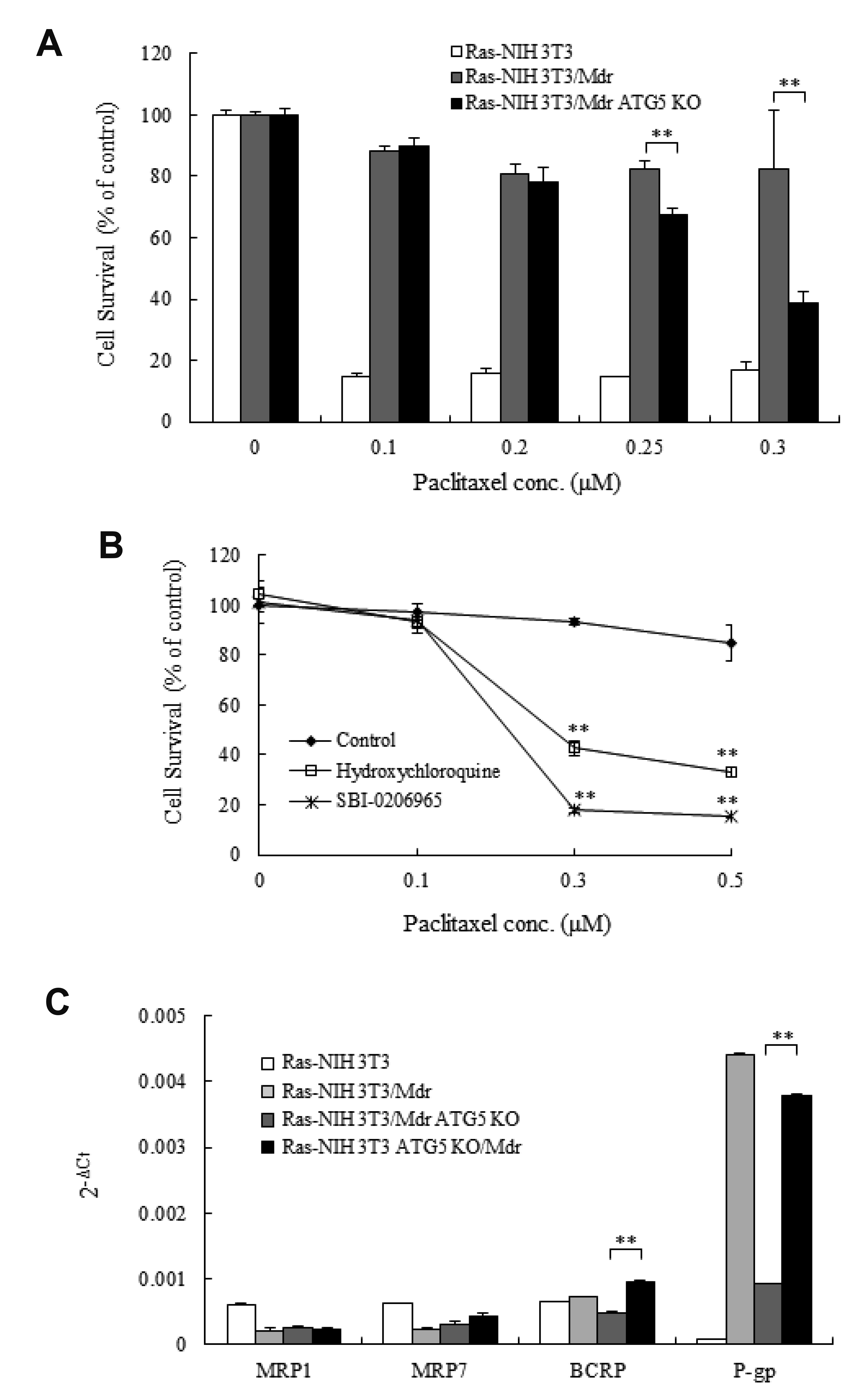 | Fig. 2Effects of an ATG5 knockout (KO) mutation on multidrug resistance.(A) Ras-NIH 3T3, Ras-NIH 3T3/Mdr and Ras-NIH 3T3/Mdr ATG5 KO cells were treated with increasing concentrations of paclitaxel ranging from 0.1 to 0.3 μM for 3 days. The cytotoxicity was evaluated using the WST-1 assay. The viability of cells treated with vehicle alone was set to 100%. Each point represents mean ± SD of quadruplicate determinations of one (of three) representative experiment. **p < 0.01 as compared with Ras-NIH 3T3/Mdr cells, as determined by Dunnett’s t-test. (B) Ras-NIH 3T3/Mdr cells were incubated in the presence and absence of SBI-0206965 (early autophagy inhibitor, 2 μM) or hydroxychloroquine (late autophagy inhibitor, 15 μM) for 3 days. The cell viability was then evaluated with the WST-1 reagent. The values represent mean ± SD of quadruplicates from one of three representative experiments. **p < 0.01 compared with control cells, as determined by Unpaired t-test. (C) Real-time RT-PCR was performed to measure the mRNA expression levels of four ABC transporters from Ras-NIH 3T3/Mdr ATG5 KO cells. The comparative threshold cycle (Ct) method was used to present relative gene expression. The expression of the target genes was normalized to β-actin expression. Each point represents mean ± SD of quadruplicate determinations of one (of three) representative experiment. MRP, multidrug resistance (MDR) protein; BCRP, breast cancer resistant protein; P-gp, P-glycoprotein. **p < 0.01 as determined by the Dunnett’s t-test compared to Ras-NIH 3T3/Mdr cells.
|
ATG5 overexpression fails to restore paclitaxel resistance in ATG5 KO cells
Next, we investigated the effect of ATG5 overexpression on paclitaxel resistance in ATG5 KO cells, previously shown to be sensitive to paclitaxel. The ATG5 overexpression was verified by western blot analysis (Fig. 3A). We observed that overexpression of ATG5 induced a shift from LC3-I to LC3-II in paclitaxel-treated ATG5 KO cells (Fig. 3B). Unexpectedly, ATG5 overexpression exerted little effect on paclitaxel resistance of ATG5 KO cells (Fig. 3C). These results raise the possibility that low level of resistance to paclitaxel in ATG5 KO cells may be related to other roles of ATG5 independent of its function in autophagy.
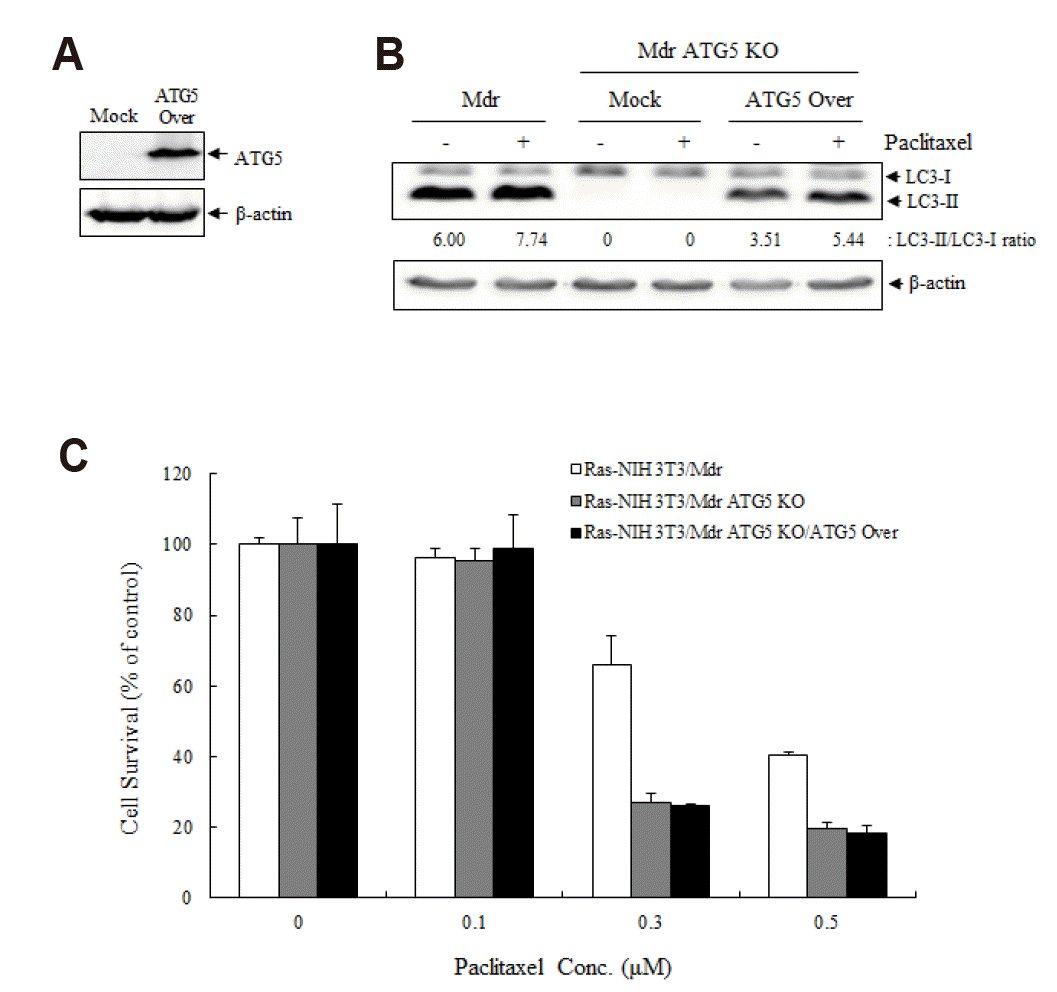 | Fig. 3The effect of ATG5 overexpression on the resistance to paclitaxel in ATG5 knockout (KO) cells.In (A–C), cells were transiently transfected either with the mock control vector or with pCI-neo-mAtg5 for 24 h. (A) Overexpression of ATG5 was confirmed by immunoblotting. (B) The conversion of LC3-I to LC3-II was identified by immunoblotting. β-Actin expression was used as a protein loading control. Results presented here are representative of at least three independent experiments. (C) The cytotoxicity was evaluated using the WST-1 assay. Each point represents mean ± SD of quadruplicate determinations of one (of three) representative experiment.
|
ATG5 KO promotes necrosis following G2/M arrest in response to paclitaxel
Flow cytometric analysis was performed to assess the cell cycle changes of ATG5 KO cells treated with paclitaxel. As shown in Fig. 4A, ATG5 KO caused a significant reduction in the cell population of cells in the S phase (18.572% in ATG5 KO versus 30.186% in their parental MDR cell populations) but an increase in the population of cells in the G2/M phase (28.676% in ATG5 KO vs. 15.703% in their parental MDR cell populations). The number of cells in the G1 phase remained almost unchanged (52.752% vs. 54.111% for ATG5 KO and their parental MDR cells, respectively). More interesting, paclitaxel caused a much stronger G2/M arrest in ATG5 KO cells (84.065%) than in their parental MDR cells (36.999%). Overexpression of ATG5 in ATG5 KO cells displayed a cell cycle distribution similar to that of mock control cells regardless of paclitaxel treatment.
 | Fig. 4Necrosis-inducing effects of paclitaxel on ATG5 knockout (KO) cells.(A) The ATG5 KO and their parental multidrug resistance (MDR) cells were treated with paclitaxel for 48 h. Cell cycle analysis was assesses using flow cytometry of propidium iodide-stained cells. Cell number and DNA fluorescence intensity are plotted on the vertical and horizontal axes, respectively. The G0/G1, S and G2/M populations were quantified using the CellQuest Pro software. Results presented here are representative of at least three independent experiments. (B) Cells were treated with paclitaxel (0.5 μM) for 48 h, stained with annexin V/propidium iodide (PI) and subjected to the flow cytometry analysis. The flow cytometry profile shows the Annexin V-FITC staining and the PI staining on the horizontal and vertical axes, respectively. The numbers shown indicate the percentage of cells in each quadrant. A group treated with camptothecin (5 μM) was used as a positive control. Results presented here are representative of at least three independent experiments. VH, vehicle; CTL, control.
|
The data presented in Fig. 4B revealed that, in ATG5 KO cells, paclitaxel promoted necrosis, in addition to apoptosis. Population of necrotic cells increased significantly from 3.53% (in parental MDR cells) to 13.04% (in ATG5 KO cells). The proportion of late apoptotic cells also increased from 0.7% to 6.88% in ATG5 KO cells compared with their parental MDR cells after treatment with paclitaxel. When camptothecin was used as a positive control for apoptosis, there was little or no difference in apoptotic cells between ATG5 KO and their parental MDR cell populations. These data suggest that the difference in the necrotic cell population may contribute to the difference in sensitivity to paclitaxel between ATG5 KO and their parental MDR cells.
Go to :
DISCUSSION
Paclitaxel is one of the most effective anticancer agents, with a wide spectrum of activity against a variety of malignant tumors [10,35]. However, tumor cells gradually develop a resistance to paclitaxel during the course of treatment [12]. A recent study reported that biphasic changes in autophagy contribute to resistance development in response to paclitaxel treatment [19]. We have also previously reported that targeting autophagy may be an effective therapeutic strategy for killing cancer cells [36]. However, it is still unclear whether autophagy protects cells from cell death or is a component of the mechanism of cell death in cells undergoing stress. Thus, we performed the additional experiments to verify whether defective autophagy could be associated with paclitaxel sensitivity in MDR cells with an ATG5 KO. Interestingly, we found that the ATG5 KO restored the sensitivity of MDR cells to paclitaxel treatment, proving that MDR cells could be re-sensitized to this drug. These results suggest that ATG5 deficiency may underlie to paclitaxel sensitivity in MDR cells. In accordance with our results, there are several reports indicating that drug sensitivity was significantly inhibited by knockdown of ATG5 [37,38]. Unexpectedly, ATG5 overexpression failed to rescue paclitaxel resistance in ATG5 KO cells despite restoring LC3 conversion. These results raise the possibility that low level of resistance to paclitaxel in ATG5 KO cells may be related to roles of ATG5 independent of its function in autophagy. However, the exact way in which ATG5 contributes to paclitaxel resistance still has to be elucidated as it could be direct or through modulation of the autophagic pathway.
A variety of resistance mechanisms have been reported; however, most studies focused on ABC transporters that may boost the efflux of chemotherapeutic agents from cancer cells [39]. Among 49 types of human ABC transporters, P-gp and MRP7 have been known to be involved in resistance to paclitaxel [14,39]. Our study revealed that the expression level of P-gp, which is increased in cells with acquired MDR, significantly decreased after an ATG5 KO. A recent study reported that paclitaxel resistance was associated with upregulated autophagy in MDR cells overexpressing P-gp [40]. However, MDR cells established from ATG5 KO cells showed a strong expression of P-gp, comparable to that of MDR cells with functional autophagy, implying that autophagy deficiency might not be the direct cause of the down-regulation of P-gp. Consistent with our hypothesis, it has been previously reported that P-gp expression was not obviously changed despite inhibition of autophagy [41]. On the other hand, the expression of MRP7 did not show correlation with paclitaxel resistance of the cells, regardless of whether they were MDR cells with functional autophagy or MDR ATG5 KO cells. Interestingly, we found that the expression of MRP1 showed inverse relation with paclitaxel resistance. This result is consistent with a study from Yan et al. [42] who reported that acquired resistance to paclitaxel resulted in downregulation of MRP1 expression. BCRP has been also recently identified as a molecular cause of MDR in diverse cancer cells [43], but BCRP seems to play little role in paclitaxel resistance [44]. In our study, we also found that BCRP expression was almost unchanged regardless of paclitaxel resistance; the ATG5 KO induced a marginal decrease in BCRP expression.
It has been reported that paclitaxel causes cell-cycle arrest at the G2/M, followed by apoptosis through microtubule disorganization [11]. In this study, we revealed that paclitaxel caused a much stronger G2/M arrest in ATG5 KO cells than in their parental MDR cells. Interestingly, paclitaxel caused a significant increase in the population of necrotic cells (7.34% in control vs. 13.04% in paclitaxel-treated group) but the population in apoptotic cells remained almost unchanged (7.34% vs. 6.88% for control and paclitaxel-treated group, respectively). Thus, our data suggest that necrosis is a major cell death mechanism induced by paclitaxel in ATG5 KO cells. These results imply that paclitaxel sensitivity in ATG5 KO cells might be associated with an increase in necrotic cell death following a marked G2/M arrest.
Overall, our findings provide ATG5 as a chemotherapeutic mechanism for the treatment of paclitaxel-resistant tumor cells. However, ATG5 overexpression restored autophagy function in ATG5 KO cells, but failed to rescue paclitaxel resistance, suggesting that increased sensitivity to paclitaxel may be related to roles of ATG5 independent of its function in autophagy. Thus, further research will be required to clarify the exact way in which ATG5 contributes to paclitaxel resistance could be direct or through modulation of the autophagic pathway.
Go to :
 XML Download
XML Download