

 PDF
PDF Citation
Citation Print
Print



INTRODUCTION
According to the revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides (CHCC) in 2012, microscopic polyangiitis (MPA) is a non-granulomatous necrotizing vasculitis with few or no immune deposits, predominantly affecting small vessels [1]. The incidence of MPA is estimated to be from 2.7 to 11.6 per million in Europe [2]. The mean age of onset is between 50 and 60 years. However, the mean age of onset in children is 12 years. Child-onset MPA shows clinical features similar to adult-onset MPA; however, unlike adult-onset MPA with no gender predilection, there is a female predominance (about 80%) in children [3,4]. In pediatric patients, renal system involvement is the most common in MPA (75%∼100%), followed by systemic, musculoskeletal, cutaneous, and lower respiratory involvement (79%, 57%, 44%, and 37%, respectively) [3,5,6].
Although the confirmation of MPA is based on pathologic findings, it is generally associated with anti-neutrophil cytoplasmic antibodies (ANCAs) and, thus, it is classified as ANCA-associated vasculitis (AAV). A recent systematic review reported that ANCAs are detected in more than 90% of cases [3]. However, a single-center study reported that 26% of 48 children with MPA were ANCA negative [7]. Thus, a negative ANCA test cannot exclude MPA.
There are no reports on the clinical characteristics of child-onset ANCA-negative MPA in Korea. So far, only two cases of ANCA-positive MPA with renal involvement in the pediatric population have been reported in Korea [8,9]. We present a rare case of ANCA-negative MPA in a 9-year-old girl with initial pulmonary manifestations.
Go to : 
CASE REPORT
A 9-year-old girl presented to the hospital with dyspnea, cough, productive of sputum, and rhinorrhea which she has had for 16 days. A few days after symptoms onset, she continued to experience worsening dyspnea and chest discomfort despite treatment with oral mucolytics and short-term prednisolone at a primary clinic. At the initial presentation to our hospital, she was noted to have coarse breathing sounds with crackles on auscultation. Her initial chest X-ray (CXR) showed peribronchial infiltration in both lungs (Figure 1A). There were no cutaneous lesions, lymph node enlargement, mucosal ulceration, arthralgia, or ophthalmological symptoms. Also, her growth and development were normal (height 50∼75 percentile, weight 10∼25 percentile, and body mass index 10∼25 percentile).
 | Figure 1Chest X-ray & chest computed tomography (CT) before lung biopsy. (A) Initial chest X-ray and CT scan: Initial chest X-ray (09/11/2016) showed peribronchial hazy infiltrations in both lower lung zones. Chest CT scan showed diffuse bronchocentric ground glass opacities and consolidation in bilateral lungs suspected to atypical pneumonia. (B) Follow-up chest X-ray and CT scan in the period of symptom worsening: Chest X-ray showed an interval improved aeration but residual streaky and granular infiltrations. Chest CT scan showed a resolution of previously noted ground glass opacities in both lungs, leaving diffuse fibro-streaky changes and some consolidations.
|
Despite a 5-day treatment course with roxithromycin in our hospital, no improvements in dyspnea and CXR findings were seen; she was thus admitted. On hospital day 1 (HD#1), her blood pressure was 98/49 mmHg, respiratory rate was 32 breaths/min, heart rate was 102 beats/min, and body temperature was 37.1°C. Her percutaneous oxygen saturation (SPO2) was 92% at room air, necessitating supplemental oxygen via nasal cannula. Her initial white blood cell count was 16,850/mm3 with 83.6% segmented neutrophils. Her initial serum chemistry was within normal limits, except for mild elevation of C-reactive protein (1.89 mg/dL). Immunoglobulin M anti-mycoplasma antibody and multiplex real-time polymerase chain reaction (PCR) for respiratory viruses were negative. Urinalysis was significant for 1+ urine albumin and microscopic hematuria. Chest computed tomography (CT) showed diffuse bronchocentric ground-glass opacity and consolidations in the lungs (Figure 1A). On HD#3, the patient was noted to have a fever and her SPO2 dropped to 88% on room air. Diagnostic bronchoscopy with bronchoscopic alveolar lavage (BAL) and fluid analysis showed 42% macrophages, 49% neutrophils, and 9% lymphocytes. No microorganism, including respiratory syncytial virus, adenovirus, influenza virus, parainfluenza virus, Epstein–Barr virus, cytomegalovirus, mycoplasma, chlamydia, legionella, fungi, and acid-fast bacilli were detected from BAL fluid following PCR or cultures. On HD#5, intravenous piperacillin/tazobactam and immunoglobulin were initiated. Although her fever subsided, her other respiratory symptoms persisted. Spirometry showed a severe restrictive pattern (FVC 22% of predicted, FEV1 22% of predicted), and Hb-corrected diffusing capacity of carbon monoxide was 21% on HD#9. Because suspicion for interstitial lung disease (ILD) was now high, we empirically started the patient on oral prednisolone at a dose of 1 mg/kg/dose twice a day [10]. After that, dyspnea gradually improved. She was discharged with a half dose of prednisolone on HD#14.
One week after discharge, her symptoms and CXR findings improved. After tapering of oral prednisolone for the next six days, she started experiencing cough, dyspnea on exertion, and pleuritic chest pain. A follow-up chest CT (Figure 1B) showed resolution of a previously noted ground-glass opacities in both lungs; however, left diffuse fibro-streaky changes and consolidation, suggestive of ILD, were noted. Serum auto-antibody screening tests for vasculitis, including antinuclear antibody, anti-basement membrane antibody, and ANCA, were all negative. A video-assisted thoracoscopic lung biopsy was performed and the biopsied specimen was suggestive of MPA, mainly involving the capillaries and small vessels with associated pulmonary hemorrhage, subpleural intra-alveolar hemorrhage, fibrin deposition, and hemosiderin-laden macrophages. Increased lymphoplasma cell and neutrophil infiltration in the interstitium were also noted (Figure 2). She was then started on oral cyclophosphamide with prednisolone twice a day for remission induction of MPA. After 12 days, her symptoms, infiltration on CXR, and pulmonary function improved. After six weeks of symptoms remission, oral azathioprine (2 mg/kg/day), instead of cyclophosphamide and low-dose prednisolone, was started for maintenance treatment. All pulmonary function parameters normalized after 6 months of treatment (Figure 3). Follow-up chest CT after 9 months did not show her previous abnormal findings. Follow-up urinalysis showed intermittent microscopic hematuria without proteinuria. After 24 months of maintenance treatment, all medications were discontinued. She has remained symptom-free for one year now based on regular exams done at the pediatric nephrology and pulmonology outpatient clinic (Figure 4).
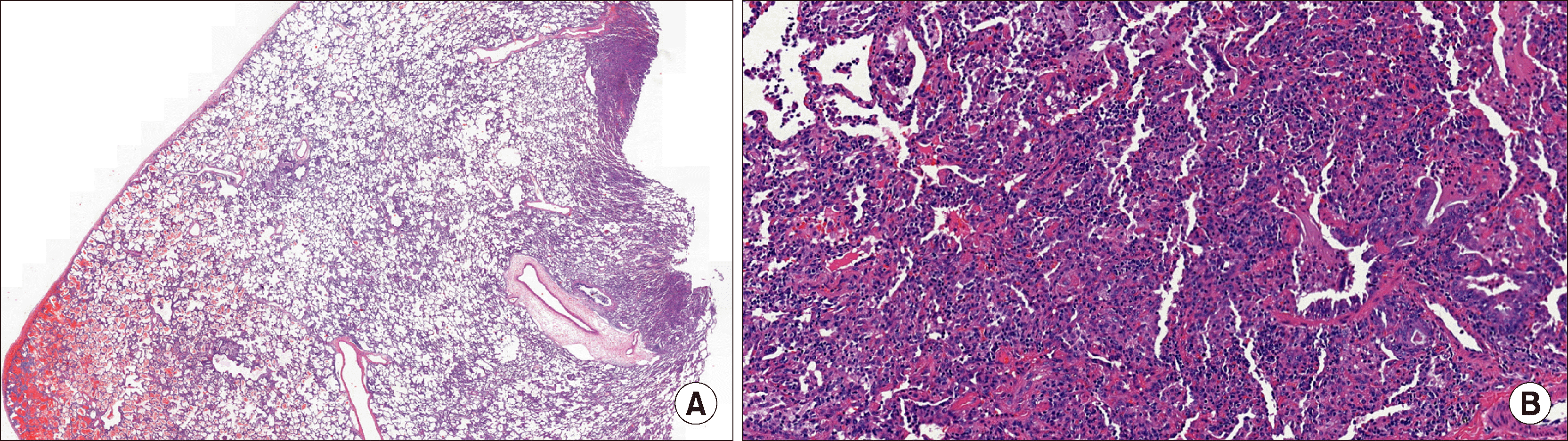 | Figure 2Pathologic findings of the biopsy specimen. (A) Surgical lung biopsy specimen shows subpleural and intraalveolar hemorrhage (H&E, ×2). (B) At higher magnification, fibrin deposition, hemosiderin-laden macrophages in alveolar spaces and capillaritis associated with neutrophils can be seen (H&E, ×100).
|
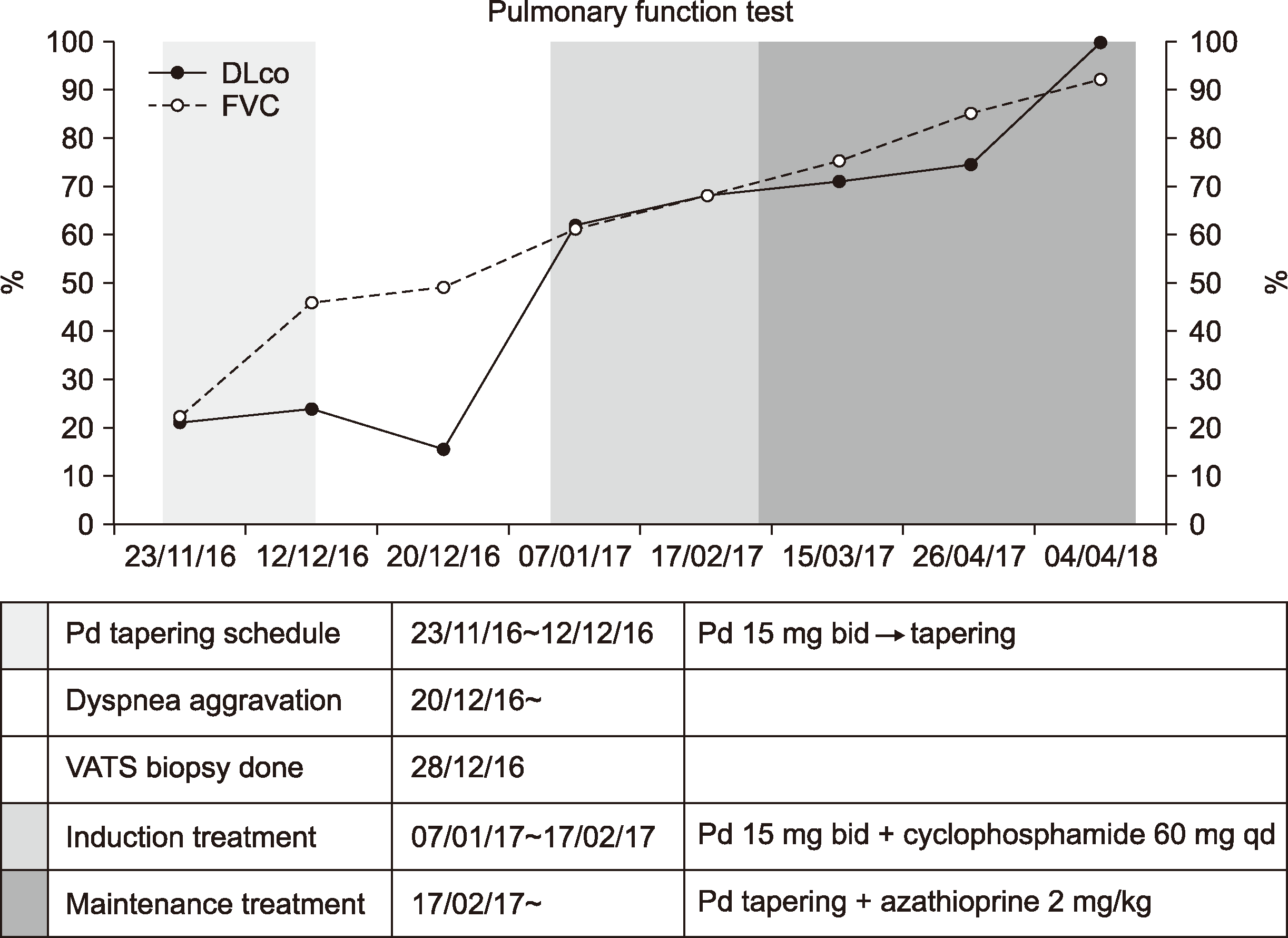 | Figure 3Changes in pulmonary function tests according to treatment. Gradual improvement of forced vital capacity and diffusion capacity is noted after remission induction treatment. DLco: carbon monoxide diffusing capacity, FVC: forced vital capacity, VATS: video-assisted thoracoscopic surgery, Pd: prednisolone, bid: twice daily, qd: once daily.
|
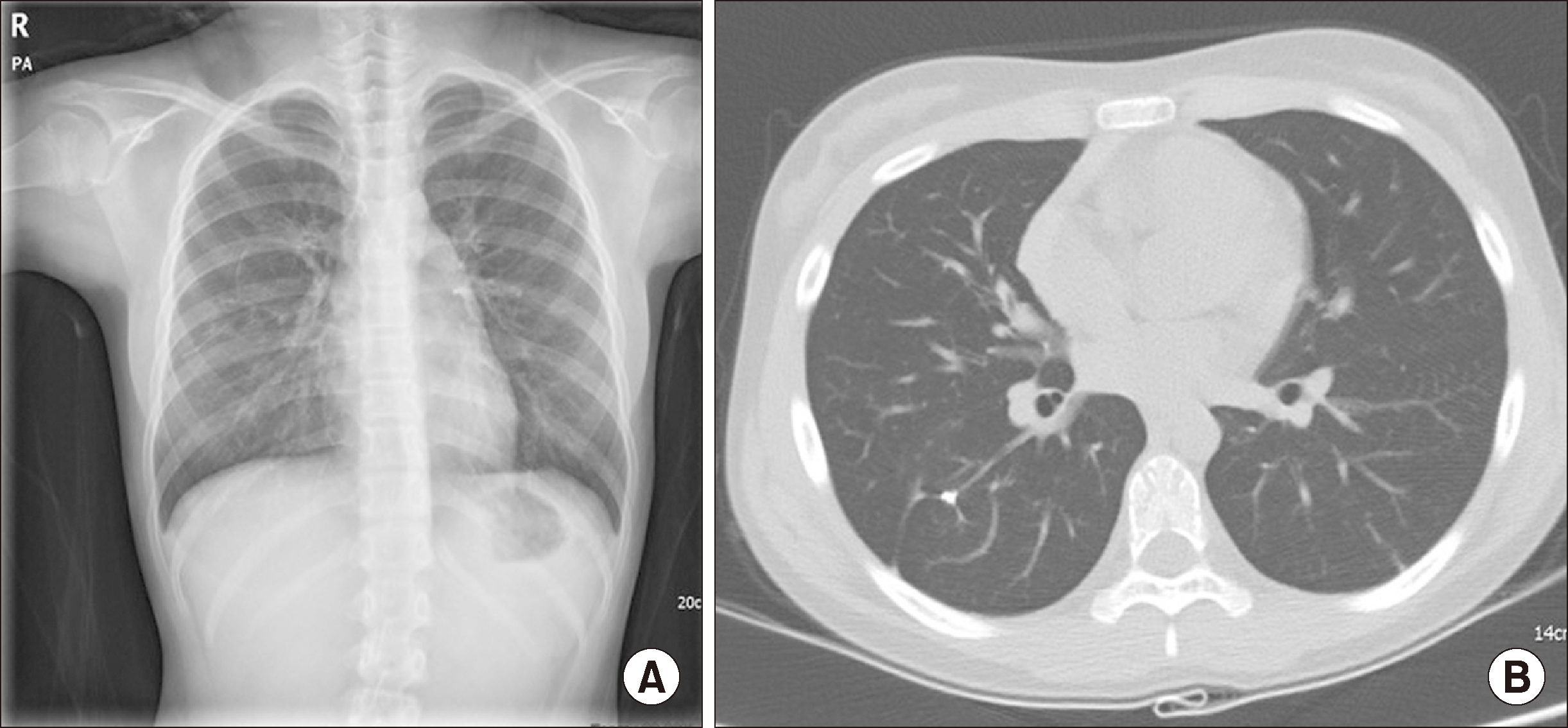 | Figure 4Chest X-ray & computed tomography (CT) scan performed at the one year after treatment. (A) Chest X-ray showed no active lung lesion except for postoperative changes in right lung. (B) Chest CT scan showed disappeared previously-noted diffuse fibrostreaky changes and consolidation in both lungs.
|
Go to :
DISCUSSION
This is the first case report on child-onset ANCA-negative MPA with initial pulmonary manifestations in Korea. Although renal or other organ involvement is frequently reported in MPA, our patient did not show any other symptoms except microscopic hematuria. In cases of ILD or an unusual course of atypical pneumonia, systemic vasculitis, including MPA, should be excluded even without multi-organ involvement. An aggressive diagnostic procedure, including lung biopsy, is needed for the precise diagnosis and treatment of MPA.
Based on the recently revised 2012 CHCC guidelines, MPA is classified as an AAV. ANCA is an important characteristic of AAV; however, a negative ANCA serum test does not exclude AAV. Currently, classifying AAVs according to the presence of ANCAs as well as their antigen specificity can convey clinically useful information more readily than a diagnosis based on clinical features alone [11]. A Korean study found that ANCA-negative patients had the youngest mean age at diagnosis among the subjects [12]. However, the differences in clinical features between seropositive and seronegative cases are controversial. A Korean study in 2017 showed no meaningful difference in cumulative relapse-free survival rate, according to MPO-ANCA, PR3-ANCA, or absent ANCA in MPA patients [12]. Another study reported that the absence of ANCA was a predictor of renal survival in a Kaplan–Meier survival analysis. However, in the multivariable Cox hazards model, the absence of ANCA did not affect cumulative renal involvement or patient survival [13].
Early diagnosis of MPA is essential because of the risk of deterioration, relapse, and adverse reactions to the medications. MPA commonly shows multi-visceral involvement. However, in the case of a single organ involvement without glomerulonephritis or alveolar hemorrhage, delayed diagnosis is possible. The pulmonary manifestations of MPA are cough, dyspnea, hemoptysis, and pleuritic chest pain caused by pulmonary capillaritis and diffuse alveolar hemorrhage. Pulmonary fibrosis is associated with a high mortality rate. If patients with atypical pneumonia or suspected ILD experience unexpected progression of their symptoms during the treatment, physicians should consider vasculitis, including MPA, as a diagnosis.
The treatment of MPA comprises two phases: remission induction and maintenance. Combinations of glucocorticoids and other immunosuppressants are commonly recommended for remission induction [14]. Glucocorticoids can be used alone as first-line therapy in patients with non-severe MPA. However, more than half of these patients eventually require the addition of another immunosuppressant because of progressive, refractory, or relapsing patterns of the disease [12]. Cyclophosphamide and rituximab are two common agents combined with glucocorticoids. These regimens can induce remission in more than 80% of patients. However, cyclophosphamide is associated with an increased frequency of neutropenia and infections, risk of infertility, and late complications such as malignancy [15]. The regimen for maintenance therapy is a low-dose glucocorticoid with azathioprine for at least two years [14]. In a randomized trial, azathioprine was effective in preventing relapse and it reduced drug toxicity related to cyclophosphamide [15].
Advances in the treatment of systemic vasculitis have significantly improved patient outcomes in the last few decades. The remission rate was up to 62% (95% confidence interval, 20%∼96%) [3]. The 5-year survival rate has been more than 70% of patients with AAV [14]. Because of the absence of controlled trials involving pediatric AAV, the treatment for child-onset MPA is still based on adult data. Since children with MPA may accumulate damage as a result of the disease and due to drug-related toxicity over time, careful observation is mandatory [6]. In our case, follow up in the outpatient after hospitalization showed intermittent microscopic hematuria without proteinuria, hypertension, or azotemia. Because renal involvement may occur in 75% to 90% of MPA patients, and it often shows a more severe course than that of pediatric GPA [14], close observation is needed.
Go to :
SUMMARY
In summary, we presented a rare case of ANCA-negative pediatric MPA initially manifesting with respiratory symptoms. Because MPA can affect multiple organs, including the lung, the differential diagnoses should be kept broad and various diagnostic approaches should be implemented when an unusual course of atypical pneumonia or ILD is encountered.
Go to :
 XML Download
XML Download