

 PDF
PDF Citation
Citation Print
Print



Introduction
Caudal regression syndrome (CRS) is a rare congenital abnormality that commonly presents with varying degrees of caudal vertebral agenesis or dysgenesis, in addition to malformations of the spinal cord [1]. There is a wide variety of associated variations in patients with CRS relating to the anatomical level of their spinal agenesis [1]. When compared with other neural tube defects, CRS patients have a much higher incidence of renal and genitourinary malformations and have a high risk of developing renal function impairment [2]. The severity of the disorder has been observed as being inversely proportional to the level of residual spinal cord function [3]. The incidence of CRS is uncommon with an estimated rate of 0.1–0.25 per 10,000 uncomplicated pregnancies [4]. In this report, we present a rare case of CRS in a patient with partial sacral agenesis and a pelvic kidney and discuss the importance of screening and early intervention.
Go to : 
Case Report
A 13-month-old female presented with chronic urinary bladder infections. The child was the product of a normal gestation and the mother was not diabetic. No cutaneous stigmata were identified on the back of the patient. The patient was walking with assistance by the parents. The lower limbs were well formed although the mass of the calf was felt to be slightly smaller than normal for age. Ultrasound images were suggestive of urinary retention. On plain abdominal X-ray, hypoplasia of the lower sacrum was observed. The S3-5 segments of the sacrum were absent (Fig. 1). A T1-weighted sagittal magnetic resonance imaging demonstrated that the spinal cord terminated at the level of the mid L1 vertebral body and the distal cord had a typical “drumstick” appearance (Fig. 2). No other intradural malformations were noted on imaging e.g., fatty infiltrated filum terminale or other forms of tethered spinal cord. The left kidney was located between the L2 to L4 vertebral bodies, and the pelvis was occupied by a large pelvic kidney (Fig. 2).
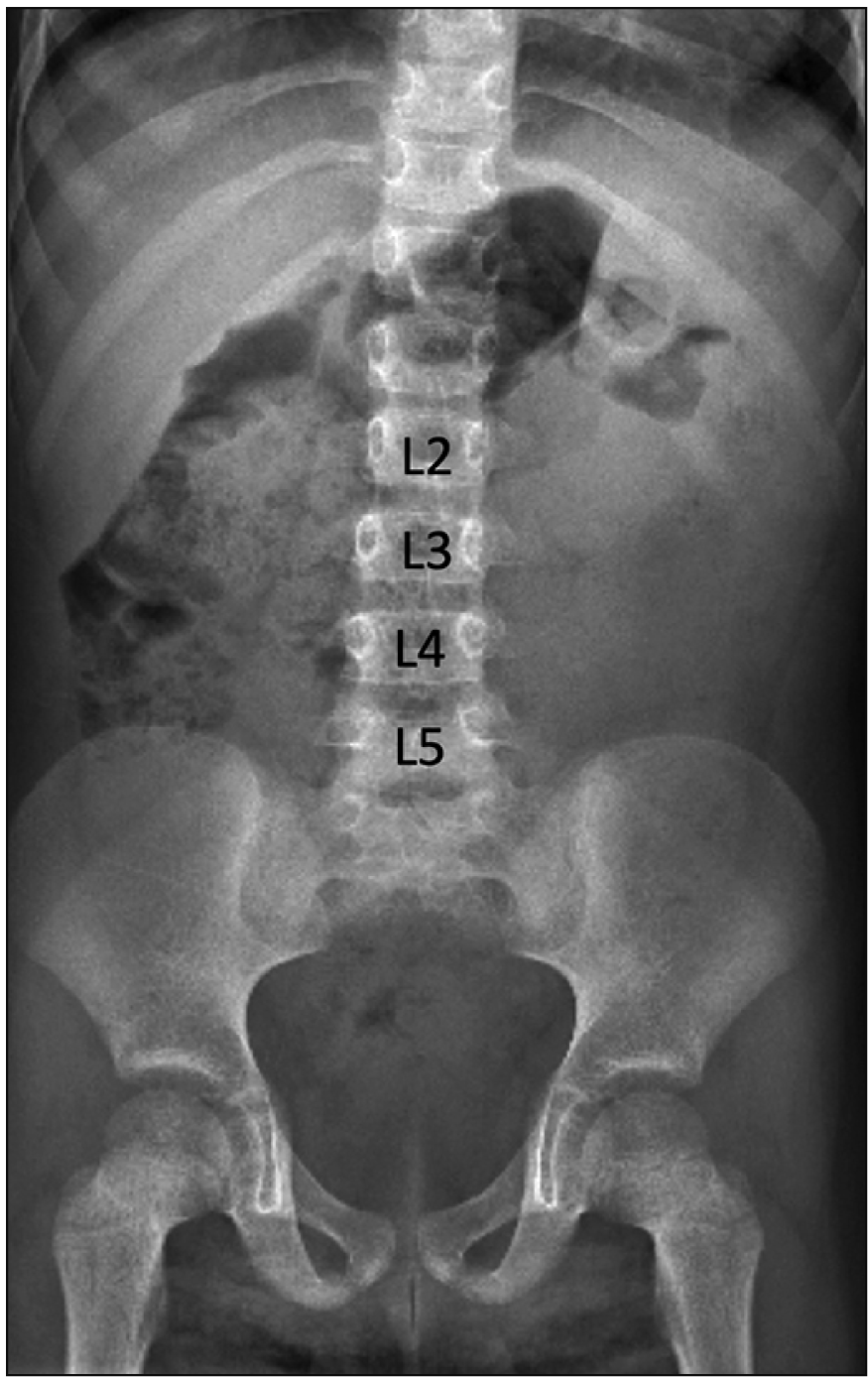
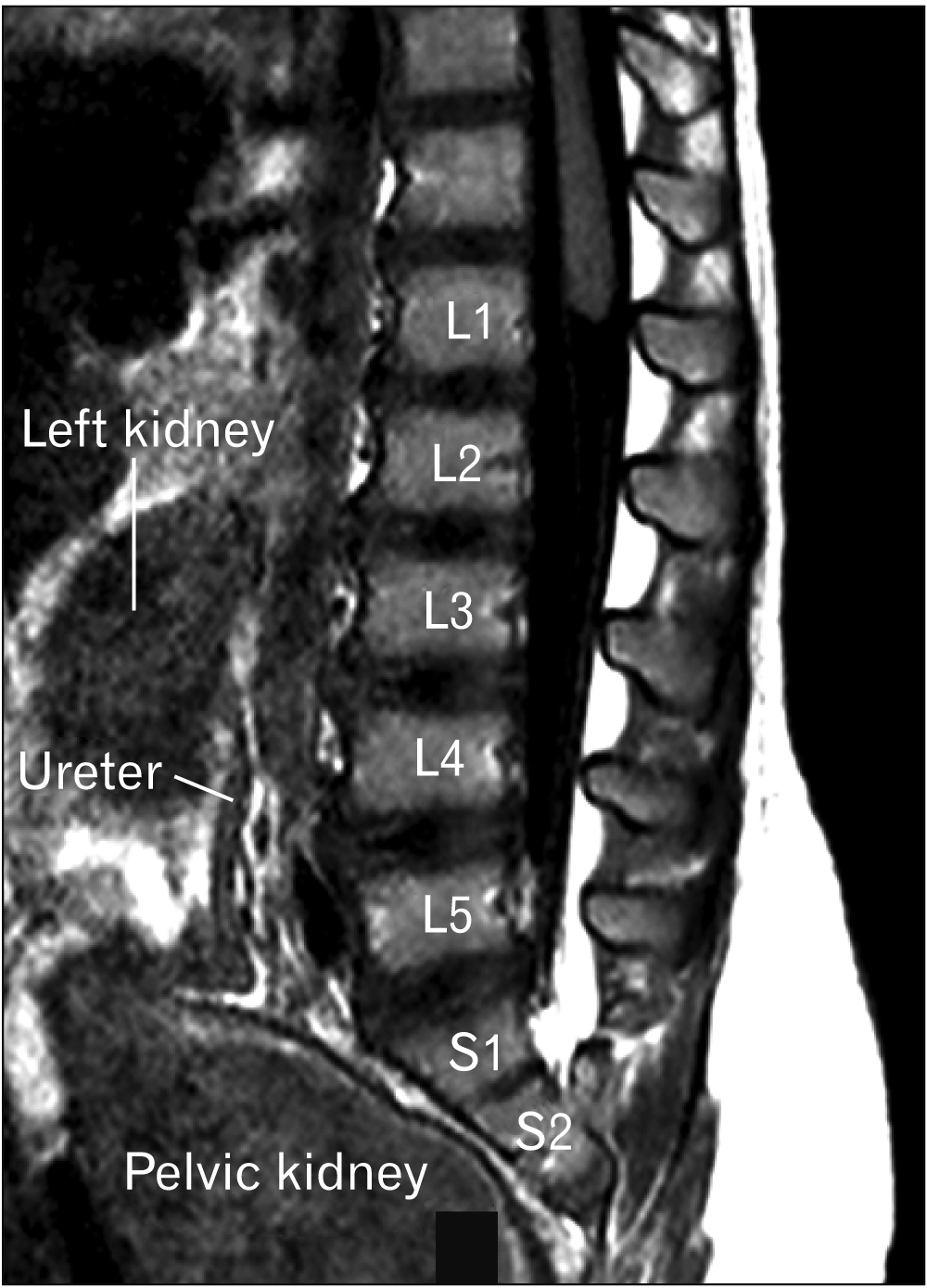
Go to :
Discussion
While the exact mechanism and etiology remain unknown, there is a significantly higher incidence associated with maternal diabetes, where 1 in every 350 births is affected [4].
Additionally, experimental studies in mice have demonstrated teratogenic effects of retinoic acid, wherein exposure of retinoic acid receptor agonists to pregnant mice induced CRS and renal anomalies in their offspring [5].
Studies conducted by Torre et al. [6] have made efforts toward quantifying the incidence of renal anomalies in patients with neural tube defects. In these studies, the most common anomalies affecting the CRS population were neurogenic bladder (60%), renal agenesis (13%–20%), and impaired renal function (8%–12%) [2, 6]. In the present case, however, the right kidney failed to ascend during the ninth week of embryogenesis and remained in the pelvis.
Pelvic kidneys are typically incidental findings as they tend to be asymptomatic [7]. There is an estimated incidence of 1 in 2,200–3,000 [8]. Even though they are not ordinarily pathogenic, they may be indicative of other conditions including nephrolithiasis, hydronephrosis, uteropelvic junction obstruction, and the development of calices [7, 8]. It has been noted that 22%–37% of patients with pelvic kidneys have also been diagnosed with uteropelvic junction obstruction [8]. Early diagnosis of uteropelvic junction obstruction is imperative as it is classified as the most common prenatally detected disease that can lead to hydronephrosis [9].
Obstructive diseases can increase the chances of infant death, kidney failure, and chronic kidney disease [9, 10]. The current hypothesis is that patients with uteropelvic junction obstruction have ureters that insert into the renal pelvis at a higher than normal location, which delays renal emptying [7, 11]. Furthermore, there is a statistically significant correlation between insufficient bladder voiding and renal damage in the CRS patient population [12]. Since the incidence of neurogenic bladder in the CRS population is 60% [2, 6], it is imperative that all CRS patients, especially those presenting with a pelvic kidney as with the present case, be thoroughly evaluated for urinary obstruction. CRS patients are often asymptomatic until their condition has progressed, therefore, utilizing a standardized system for scoring dysfunctional voiding symptoms may be the best method for assessing these patients [13]. This testing may also aid with understanding the underlying neurological issue with the spinal cord [12]. Although CRS patients have neuromuscular challenges, they have the capacity to live fairly normal lives. Thus, early urologic testing and intervention are necessary to lower their risk of developing chronic conditions that will impact their quality of life [12].
In conclusion, although renal anatomy variants are found in patients with CRS, a pelvic kidney is normally not part of the constellation of findings in this malformation. As seen in our patient, a pelvic kidney should be considered in patients suspected of having CRS.
Go to :
 XML Download
XML Download