

 PDF
PDF Citation
Citation Print
Print



Introduction
Burkitt’s lymphoma (BL) is a highly aggressive and invasive B-cell non-Hodgkin lymphoma with varying patterns of clinical behavior [1]. BL patients are slowly decreasing and cure rates also improved. However, elder patients still have poor prognosis [2]. BL is associated with Epstein-Barr virus (EBV) infection and shows clinical symptoms that have been categorized into sporadic, endemic, and immunodeficiency-associated subtypes. BL is characterized by translocations of chromosomes 8 and 14 that result in upregulation of the c-myc protein and subsequent increase of cell proliferation [3, 4]. With recent improvements in chemotherapy regimens and the application of new targeted therapies, the chance for successful treatment of children with BL has significantly improved [5, 6]. However, elderly patients and patients with advanced disease often show invasion to the central nervous system, and the prognosis for these patients is poor [7-9]. Therefore, the identification of an effective new treatment method for BL is urgently required. The development of a molecular-targeted therapy has presently taken center stage as the preferred treatment option.
Raji cells are derived from the B-lymphocytes of a male BL patient and categorized as lymphoblast-like. Some characteristics of Raji cells include a lack of differentiation, illustrated by the formation of large aggregations of hundreds of individual cells. Raji cells produce an unusual strain of EBV, which both transforms cord blood lymphocytes and induces early antigens in the cells [10, 11]. Ramos cells originated from a human Caucasian BL male patient, and are EBV-genome-negative [12]. In the present study, these two cell lines were used as representative cell lines for BL.
EBV is a herpes virus type 4 which contains about 172 kilobase double strand DNA genome encoding about 100 products and non-coding RNAs. Latent viral proteins such as latent membrane proteins (LMPs) are associated with latent program in a host and deeply related to intracellular signal transduction, cellular regulation and cell proliferation of host cells as well as maintaining EBV genome [13].
Reactive oxygen species (ROS) plays an important role in various cell functions including the process of virus-induced malignant transformation [14, 15]. A recent study reported that the increase in ROS level by EBV infection provides a growth advantage to EBV-infected cells by inducing the expression of cell proliferation-associated genes [15]. Regarding the role of ROS in B cell lymphoma including BL, another study reported that proliferation was induced through ROS increase [16]. Therefore, ROS is a potential therapeutic target in virus-associated cancers including BL. LMP1 regulates the expression Nox and Nox regulatory subunit p22phox [17].
The nicotinamide adenine dinucleotide phosphate (NADPH) oxidase (Nox) family is composed of the Nox1, -2, -3, -4, -5 and dual oxidase 1, -2 proteins and includes the major enzymes responsible for the production of ROS [18, 19]. Over-expression of Nox isoform enzymes was recently reported in colorectal cancer and non-small cell lung cancer [20, 21]. Furthermore, these enzymes play an important role in cell cycle regulation and have been proposed as a potential target in BL [22, 23]. However, the effects of the specific inhibitors for Nox isoforms and the signaling mechanisms remain poorly understood.
The Bcl-2 family proteins are major regulators of apoptosis [24, 25]. The pro-survival members (Bcl-2, Bcl-xL, Mcl-1, and A1) are integral for cell survival; the BH3-only proteins (Bim, Puma, Bad, Noxa) initiate apoptosis signaling; and Bax/Bak are required for mitochondrial outer membrane permeabilization [25-27]. The conformational changes of Bax and Bak lead to homo-oligomerization of the proteins and the formation of outer-mitochondrial membrane spanning pores [28]. These Bax and Bak pores permit the release of cytochrome c (Cyt c) into the cytoplasm, which triggers activation of the caspase cascade, and ultimately results in apoptosis [29, 30].
In this study, we evaluated the expressions of Nox isoforms and the effects and cell signaling mechanism of a specific Nox inhibitor in EBV-positive Raji BL cells in comparison with EBV-negative Ramos BL cells.
Go to : 
Materials and Methods
Cells and cell culture conditions
Peripheral blood mononuclear cells (PBMCs) were isolated from whole blood by Ficoll-paque gradient centrifugation (Amersham Life Science, Buckinghamshire, UK). Primary B cells were purified from PBMCs using a magnetic-activated cell sorting B cell-negative depletion kit (Miltenyi Biotec, Bergisch Gladbach, Germany). Ramos, an EBV-negative B lymphoma cell line, and Raji, an EBV-positive B lymphoma cell line, were obtained from the American Type Culture Collection (Rockville, MD, USA). Both cell lines were maintained in RPMI-1640 medium (HyClone, Logan, UT, USA) containing 10% fetal bovine serum (HyClone) and antibiotics at 37°C in humiditied 5% CO2 atmosphere.
Reagents and antibodies
Western blot was performed using the following primary antibodies: anti-human Nox2, anti-human Nox4 (BD Biosciences, San Diego, CA, USA), and anti-human β-actin antibodies (Santa Cruz Biotechnology, Santa Cruz, CA, USA). GSK2795039, a Nox2 inhibitor, was obtained from MedChemExpress (Monmouth Junction, NJ, USA). Anti-human caspase 3 and 9 antibodies (Abcam, Cambridge, MA, USA) and anti-human Mcl-1, Bcl-2, Bim, Noxa antibodies (Cell Signaling Technology, Beverly, MA, USA) were used to evaluate protein expression. N-Benzyloxycarbonyl-Val-Ala-Asp (O-Me) fluoromethyl ketone (Z-VAD-FMK, Sigma, St. Louis, USA), a pan-caspase inhibitor; N-acetyl-L-cysteine (NAC, Cell Signaling Technology), a ROS scavenger; and ABT-263 (Selleck Chemical, Shanghai, China), a Bim specific inhibitor were used to identify the intra-cellular signaling pathway.
Reverse-transcription polymerase chain reaction
Cells were collected and washed three times with phosphate-buffered saline (PBS, pH=7.4). Total RNA was isolated using the RNeasy Mini Kit (Qiagen, Hilden, Germany). RNA was transcribed into cDNA using the reverse-transcription PreMix Kit (Bioneer, Daejeon, Korea). Polymerase chain reaction (PCR) amplification was performed using primer sets (Bioneer) specific for Nox-2 (Forward: 5’-CAT GTT TCT GTA TCT CTG TGA-3’; Reverse: 5’-GTG AGG TAG ATG TTG TAG CT-3’) and Nox-4 (Forward: 5’-CCA TGG CTG TGT CCT GGA GGA GCT G-3’; Reverse: 5’-AGT TGA GGG CAT TCA CCA GAT GGG C-3’). For the control, a specific primer set for β-actin (Forward: 5’-ATC CAC GAA ACT ACC TTC AA-3’; Reverse: 5’-ATC CAC ACG GAG TAC TTG C-3’) was used, which yielded a 200-bp product. PCR products were visualized on 2.5% agarose gels with ethidium bromide. The normalization and fold-changes of levels of Nox2 and Nox4 expression were calculated with Image J 1.38 software (http://rsweb.nih.gov/ij/index.html). Each experiment was repeated at least three times.
Western blotting
Cells were harvested and washed twice with PBS. Cells were lysed in radioimmunoprecipitation assay (RIPA) buffer (Elpis Biotech, Daejeon, Korea) containing a protease inhibitor cocktail (AEBSF, aprotinin, bestatin hydrochloride, E-64, EDTA, and leupeptin hemisulfate salt; Sigma). To evaluate phosphorylation events, an additional set of phosphatase inhibitors were added to the RIPA buffer (Cocktail II; sodium orthovanadate, sodium molybdate, sodium tartrate, and imidazole; Sigma). Protein concentration was estimated with the bicinochoninic acid Protein Assay Kit (Pierce, Rockford, IL, USA). Equal amounts of proteins (40 μg) were separated by electrophoresis on sodium dodecyl sulfate-polyacrylamide gels and transferred to polyvinylidene difluoride membranes (Amersham) by immunoblotting. The membranes were then incubated overnight at 4°C in a PBS solution supplemented with 5% non-fat dry milk. The blots were probed with specific primary antibodies for 1 hour, incubated with diluted enzyme-linked secondary antibody, and then visualized by enhanced chemiluminescence as recommended by the manufacturer (Amersham). Equivalent protein loading was assessed by β-actin expression. Each experiment was repeated at least three times.
Evaluation of cell viability
Cells were seeded and then treated with GSK2795039 at the indicated doses for 48 hours, and cell viability was analyzed using a WST-1 assay (Sigma) or trypan blue exclusion method.
Detection of apoptosis by Annexin V/propidium iodide
Cells were harvested, washed twice with PBS and resuspended in 100 ml Annexin V binding buffer (10 mM HEPES, 0.14 M NaCl, and 0.25 mM CaCl2). Next, 2 ml FITC-conjugated Annexin V (BD Biosciences) and 1 ml PI (BD Biosciences) were added, and cells were incubated at room temperature for 15 minutes in the dark with gentle vortexing. Next, 400 ml of Annexin V binding buffer was added to each tube, and cells were analyzed using a flow cytometry.
Measurement of cytochrome c and Bax translocation
Mitochondrial and cytosolic fractions of cells were prepared using a Cytosol/Mitochondria fractionation kit (Calbiochem, San Diego, CA, USA). Briefly, cells were harvested by centrifugation at 600×g for 5 minutes at 4°C and washed twice with cold PBS. Cells were then resuspended in 250 ml cytosol extraction buffer containing protease inhibitor mixture (included in the fractionation kit) and 1 mM dithiothreitol (DTT). After incubation on ice for 10 minutes, cells were homogenized using a Dounce tissue homogenizer on ice. Homogenates were centrifuged at 700×g for 10 minutes at 4°C, and the supernatant was centrifuged again at 10,000×g for 30 minutes at 4°C. The resulting supernatants were harvested and designated as cytosolic fractions. The pellets were resuspended in 50 ml mitochondria extraction buffer containing protease inhibitor mixture and DTT and designated as mitochondrial fractions. The fractionated cell lysates (10 mg per well) were prepared for western blotting using anti-Cox IV and anti-beta tubulin for loading control.
Detection of sub-G1 level
To detect the sub-G1 peak, PI staining was performed (BD Biosciences). Briefly, cells were incubated with Nox2 inhibitor, z-VAD, NAC, or Bim inhibitor in culture medium. Cells were collected and washed according to the instructions supplied by the manufacturer. Cell pellets were stained with PI staining solution containing RNase A (10 mg/ml; Sigma) and PI (2 mg/ml) in PBS. The cell suspension was then incubated in the dark at room temperature for 20 minutes. DNA content was determined using a FACSCalibur flow cytometer (BD Biosciences).
Statistical analysis
All data are expressed as mean±standard deviation and each value represents at least two different experiments. Comparisons between all individual data were made using one-way ANOVA. Statistical significance was defined by a P-value<0.05.
Go to :
Results
Nox2 and Nox4 were highly expressed in Raji cells compared with primary B and Ramos cells
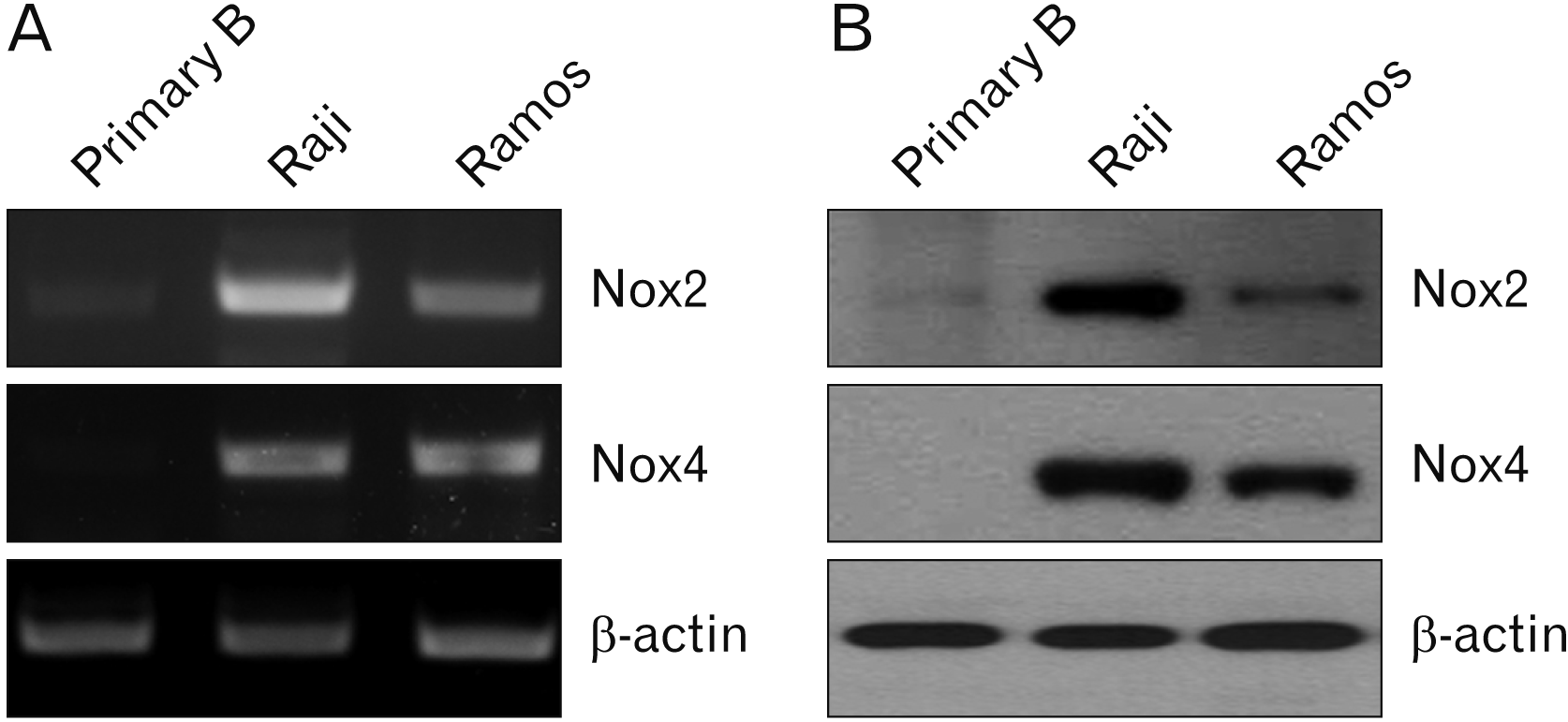
Raji is an EBV-positive B lymphoma cell line, and Ramos is an Epstein-Barr-negative B lymphoma cell line. Recent studies reported that these two cell lines showed different characteristics including phenotypes [9-11]. Nox2 and Nox4 expression were first evaluated in primary B, Raji, and Ramous cells. PCR (Fig. 1A) and western blotting (Fig. 1B) results showed that Nox2 mRNA and protein were highly expressed in Raji cells compared with the other cell lines. However, Nox4 protein only was highly expressed in Raji cells compared with the other cells.
 | Fig. 1Cell viability changes at 48 hours after Nox2 inhibitor, GSK2795039, treatment. Cells were seeded and treated with GSK2795039 at the indicated doses for 48 hours. Cell viability was analyzed using a WST-1 assay or trypan blue exclusion method. (A) Primary B cells, (B) Raji cells, (C) Ramos cells. Nox2, nicotinamide adenine dinucleotide phosphate oxidase 2; Primary B, primary B cells. The data shown are representative of three independent experiments (*P<0.05 vs. control, **P<0.01 vs. control).
|
Nox2 inhibitor treatment induced cell death in Raji cells
Our results showed that Raji cells significantly expressed Nox2 mRNA and protein as evaluated by PCR and western blotting. To assess the role of Nox2, the cellular responses of Raji cells were observed after treatment using a specific inhibitor for Nox2, GSK2795039. The Nox 2 inhibitor significantly induced cell death in only Raji cells in a dose-dependent manner. The most effective concentration of GSK2795039 was 12.5 μM concentration, and Raji cells showed the similar effects in higher concentration (Fig. 2B). GSK2795039 had no effect on primary B cells (Fig. 2A) and induced slight cell death in Ramos cells only at a high concentration; however, the induction of cell death was not statistically significant (Fig. 2C).
 | Fig. 2PCR and western blot analyses of NADPH oxidase expression in primary B, Raji, and Ramos cells. (A) NADPH oxidase mRNA expression was verified by RT-PCR. β-actin was used as a loading control. Cells were harvested, and PCR amplification was performed as described as in the Materials and Methods. (B) Cells were harvested and cell lysates were extracted. The cell lysates were subjected to western blot using anti-human Nox2, -4, and β-actin antibodies. β-actin was used as a loading control. Results are representative of three independent experiments. NADPH, nicotinamide adenine dinucleotide phosphate; Nox2, NADPH oxidase 2; Nox4, NADPH oxidase 4; PCR, polymerase chain reaction; Primary B, primary B cells; RT, reverse-transcription.
|
Nox2 inhibitor induced apoptosis in Raji cells in time- and dose-dependent manners
Our results showed that Nox2 inhibitor effectively induced cell death in only Raji cells. We next examined whether Nox2-induced cell death was associated with apoptosis. Cells treated with Nox2 inhibitor were stained with FITC-labeled Annexin V and PI and analyzed by flow cytometry. Nox2 inhibitor treatment induced the apoptosis of Raji cells in a time-dependent manner (Fig. 3). Nox2 inhibitor treatment also induced apoptotic cell death in Raji cells in a dose-dependent manner, but no effects were observed in Ramos cells. The difference in Nox2 inhibitor induced-apoptosis between Raji and Ramos cells were statistically significant at 6.25 and 12.5 μM concentrations (Fig. 4A).
 | Fig. 3Apoptosis analysis of Raji cells after Nox2 inhibitor, GSK2795039 treatment. Raji cells were treated with GSK2795039 for 48 hours. Cells were harvested, washed, and stained with FITC-conjugated Annexin V and PI as described in the Materials and Methods. Stained cells were analyzed by flow cytometry. The numbers in box represents the cell proportion in each quadrant. Data are representative of the three independent experiments. DMSO, control for GSK2795039; Negative, negative control; Nox2, nicotinamide adenine dinucleotide phosphate oxidase 2; PI, propidium iodide.
|
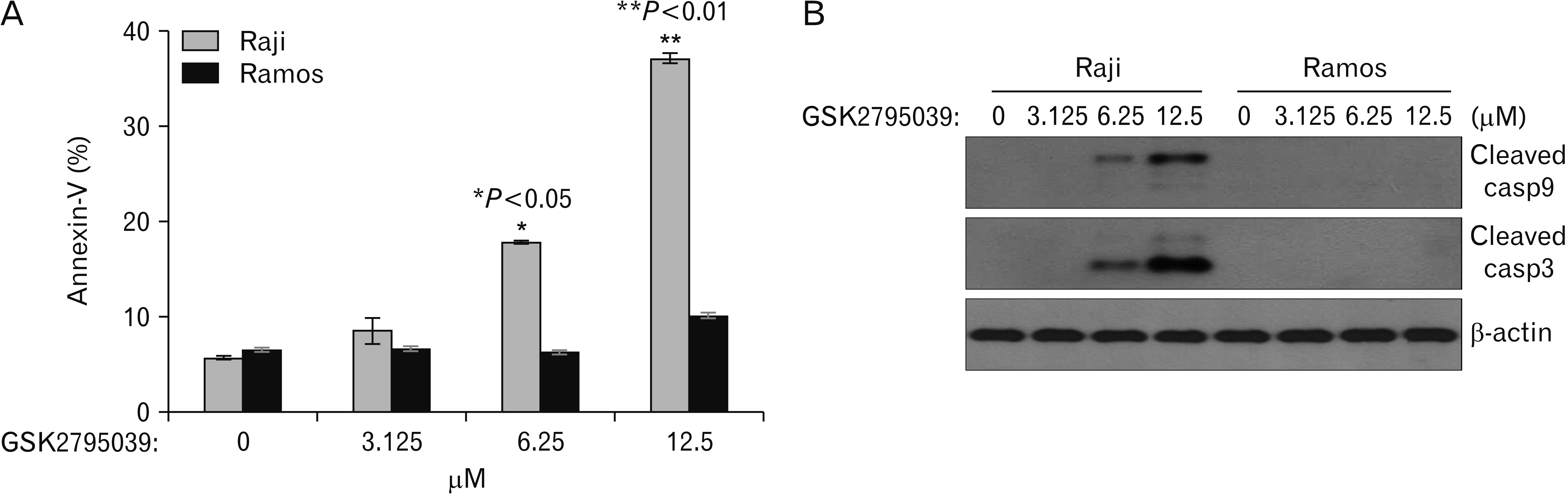 | Fig. 4GSK2795039, the Nox2 inhibitor, mediated caspase-dependent apoptotic cell death in Raji cells in a dose-dependent manner. (A) Raji and Ramos cells were treated with GSK2795039 in various concentrations for 48 hours and then stained with Annexin V-FITC. Apoptotic cell populations were determined with flow cytometry. * and ** indicate P<0.05 and P<0.01, respectively, compared with the Ramos cell apoptotic population. (B) Western blot analysis of caspase activation in cells after GSK2795039 treatment. β-actin was used as a loading control. Cell graphs represent the mean±standard deviation of three independent experiments in triplicate. Red box represents meaningful changes of molecule expressions. Nox2, nicotinamide adenine dinucleotide phosphate oxidase 2.
|
Caspases are involved in Nox2 inhibitor-induced apoptosis
Our results showed that the Nox2 inhibitor induced apoptosis in Raji cells. Caspases are key effector molecules that induce apoptosis in cancer cells [14]. Therefore, we next evaluated the activation of caspases after Nox2 inhibitor treatment in Raji cells. The results showed that the Nox2 inhibitor induced the cleavage of caspases 3 and 9 (activated forms) in Raji cells in a dose-dependent manner, but no effects were observed in Ramos cells (Fig. 4B).
Mcl-1, Bim, and Noxa are involved in Nox2 inhibitor induced-apoptosis of Raji cells
To identify the mechanism of Nox2 inhibitor-induced apoptosis, several candidate signaling molecules were evaluated by western blot. Mcl-1, Bcl-2, Bim, and Noxa were expressed constitutively in both Raji and Ramos cells; GSK2795039 treatment decreased Mcl-1 expression and increased Bim and Noxa expression in Raji cells in a dose-dependent manner, however, GSK2795039 had no effect on Ramos cells, similar to the previous results (Fig. 5).
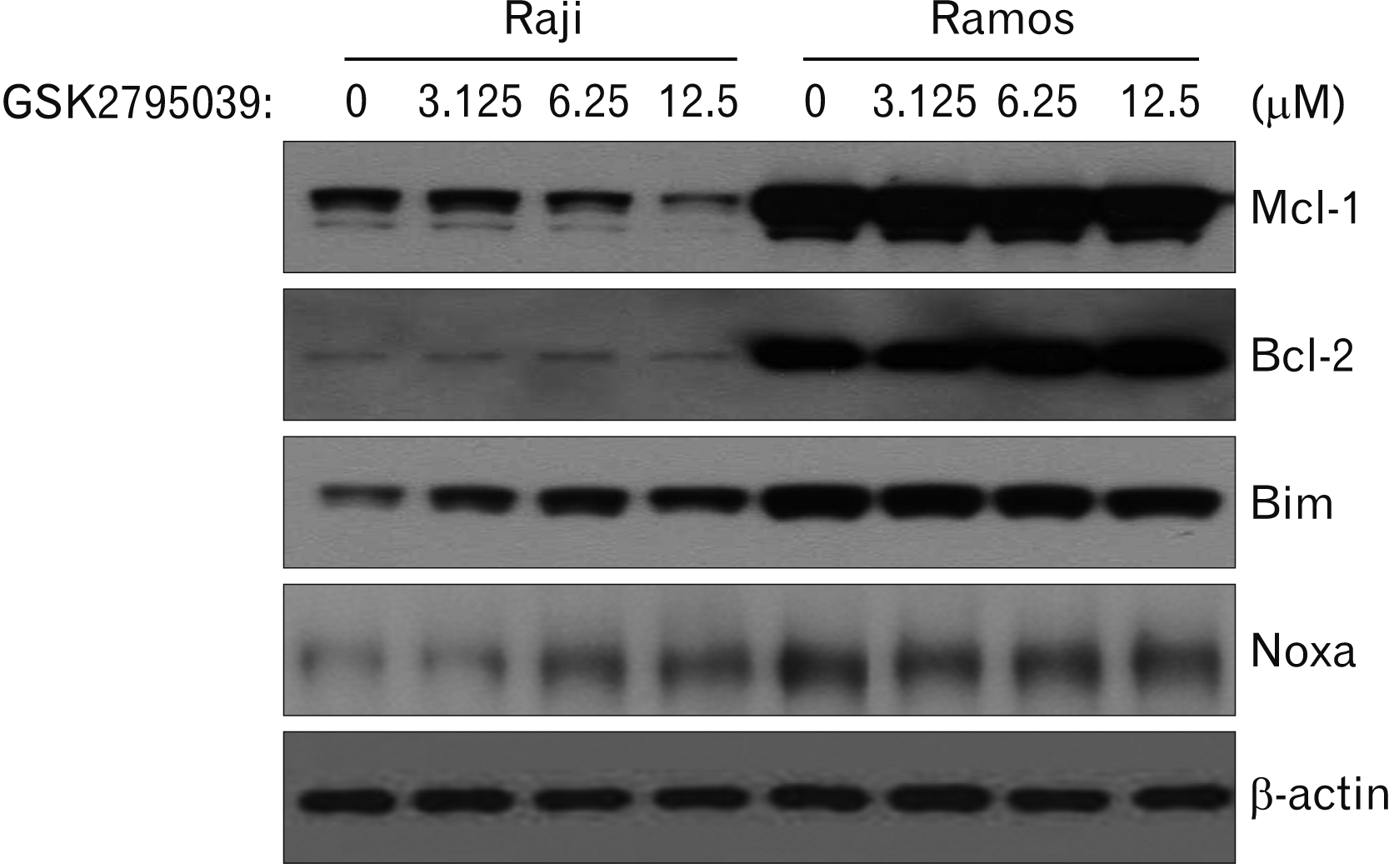 | Fig. 5Effects of the Nox inhibitor, GSK2795039, on Bcl-2 family proteins expressions in Raji and Ramos cells. After GSK2795039 treatment, cells were harvested and cell lysates were extracted. The cell lysates were subjected to western blot using anti-human Mcl-1, Bcl-2, Bim, Noxa, and β-actin antibodies. β-actin was used as a loading control. Results are representative of three independent experiments. Red box represents meaningful changes of molecule expressions.
|
Translocation of Bax and release of cytochrome c in Raji cells after GSK2795039 treatment
Next, we examined translocation of Bax, a pro-apoptotic protein, from the cytoplasm to mitochondria and release of Cyt c from the mitochondria into the cytoplasm after GSK2795039 treatment. Previous studies showed that the activation and translocation of Bim is associated with the translocation of Bax to mitochondria, and translocated Bax activates release of Cyt c, a key effector protein related with apoptosis [27, 28]. GSK2795039 treatment induced the translocation of Bax from the cytoplasm to mitochondria and release of Cyt c from mitochondria into cytoplasm (Fig. 6).
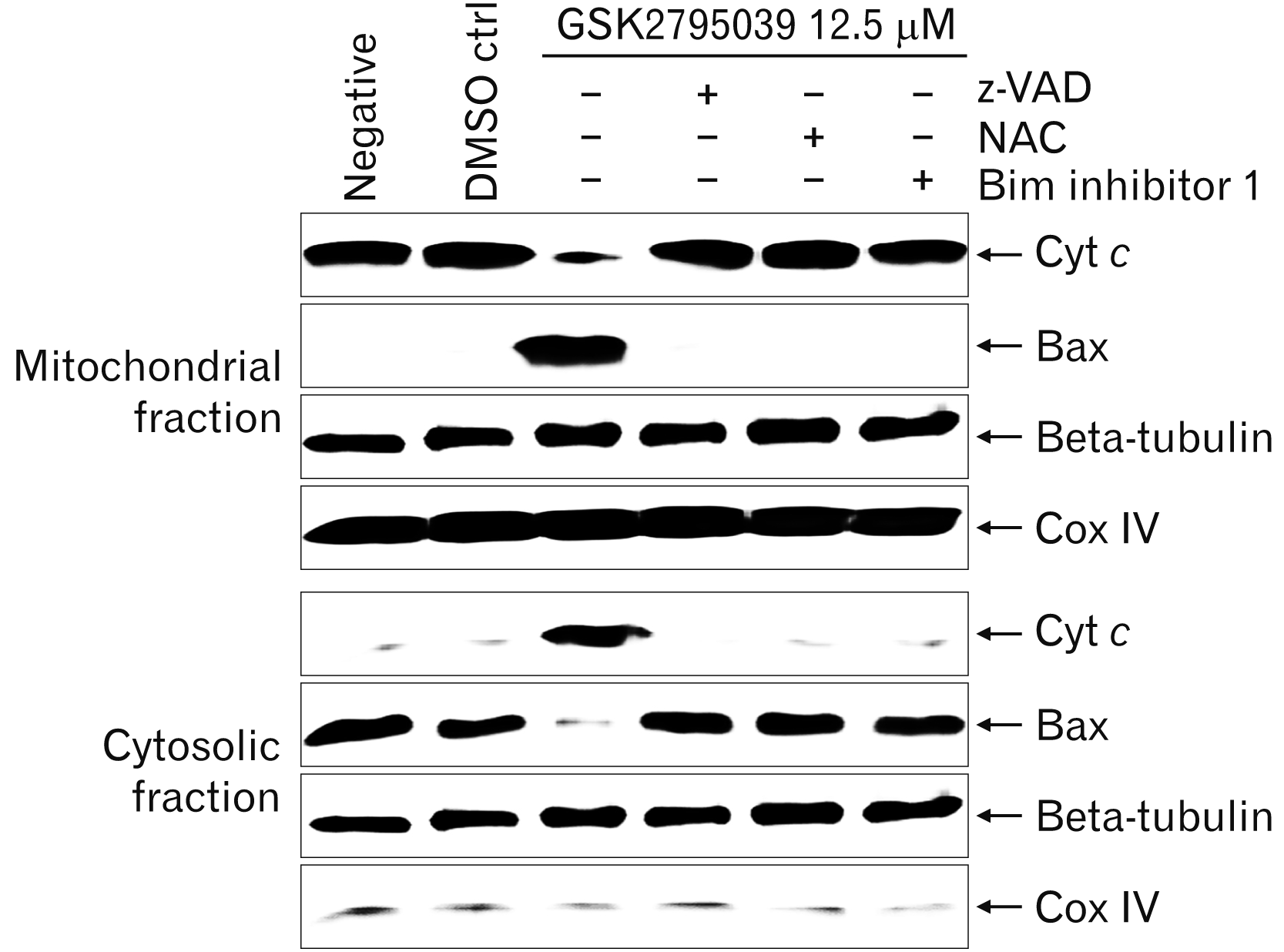 | Fig. 6Effects of GSK2795039 on the translocation of Bax and release of Cyt c in Raji cells. After GSK2795039 treatment, cells were harvested and cell lysates were extracted. The cell lysates were separated into cytosolic and mitochondrial fractions, which were then analyzed by western blotting using anti-human Cyt c, bax, beta-tubulin, and Cox IV antibodies. Anti-Cox IV and anti-beta tubulin antibodies were used for fraction loading controls. Results are representative of three independent experiments. Cyt c, cytochrome c; DMSO ctr, solvent control; GSK2795039, Nox2 inhibitor; NAC, N-acetyl-L-cysteine, reactive oxygen species scavenger; Negative, negative control without treatment; z-VAD, N-benzyloxycarbonyl-Val-Ala-Asp, a pan-caspase inhibitor.
|
Inhibition assay using z-VAD, NAC, and BI-1 in Raji cells after GSK2795039 treatment
To clarify the signaling pathway of Nox2 inhibitor induced-apoptosis in Raji cells, inhibition assays were performed using the pan-caspase inhibitor z-VAD, the ROS scavenger NAC, and the Bim inhibitor 1 BI-1. z-VAD, NAC, and BI-1 effectively blocked the translocation of Bax and release of Cyt c in Raji cells after GSK2795039 treatment (Fig. 6). Moreover, all three inhibitors completely restored the cell cycle arrest (subG1 phase in apoptotic cells) induced by GSK2795039 treatment (Fig. 7).
 | Fig. 7Inhibitory effects of z-VAD, NAC, and Bim inhibitor 1 on the changes of sub-G1 populations induced by GSK2795039 in Raji cells. To assess apoptotic cells, cells were stained with PI as described in the Materials and Methods. Cells were then analyzed by flow cytometry. The percentage represents the fraction of sub-G1 cells (M1) relative to the total number of cells. Results are representative of three independent experiments. DMSO ctr, solvent control; GSK2795039, Nox2 inhibitor; NAC, N-acetyl-L-cysteine, reactive oxygen species scavenger; Negative, negative control without treatment; z-VAD, N-benzyloxycarbonyl-Val-Ala-Asp, pan-caspase inhibitor.
|
Go to :
Discussion
Several previous studies have demonstrated that tumor virus infection is associated with increased oxidative stress through Nox induction, which exerts an important role in virus-induced transformation [29-31]. A recent study reported that EBV increases the proliferation of EBV-infected cells through the induction of ROS by cellular growth-associated factors in B lymphocytes [14].
In this study, the relationship between ROS-generating enzymes and EBV infection was assessed in two BL cell lines. We first investigated whether EBV infection could regulate the expression of the ROS-generating Nox enzyme family using EBV-positive Raji and EBV-negative Ramos BL cells. Our results showed that both Nox2 and Nox4 were highly expressed in EBV-positive Raji cells compared with EBV-negative Ramos cells. However, Nox2 was more highly expressed than Nox4 in Raji cells as shown by PCR and western blotting. Moreover, the Nox2 specific inhibitor GSK2795039 induced cell death only in Raji cells in a dose-dependent manner. The most effective concentration of GSK2795039 was 12.5 μM concentration, therefore this concentration was used in the further experiments. These results suggested that Nox2 expression depends on EBV infection and could be a possible target for treatment in EBV-positive BL tumors. A recent study reported that the EBV oncoprotein LMP1 regulates the expression of Nox and a Nox regulatory subunit [32]. This report also supports the relationship between Nox enzymes and EBV infection.
We next evaluated whether apoptosis was involved in the cell death of Raji cells by GSK2795039 treatment using an Annexin-V/PI assay kit. GSK2795039 treatment induced the apoptosis of Raji cells in time- and dose-dependent manners, but no effect was observed in Ramos cells. In addition, GSK2795039 treatment induced the activation of caspase 3 and 9 in Raji cells in a dose-dependent manner. The pan-caspase inhibitor z-VAD completely inhibited Nox inhibitor-induced apoptosis of Raji cells. These results indicated that GSK2795039 treatment induced the apoptosis of Raji cells in a caspase-dependent pathway.
Caspase activation is regulated at the multi-phase, including the release of other proteolytic enzymes such as Cyt c from the mitochondria [33]. Moreover, Bcl-2 family proteins such as Bcl-2, Bcl-xL, Mcl-1, Bim, Puma, Noxa, Bad, and Bax regulate the release of apoptosis-associated proteins from mitochondria [34, 35]. Therefore, the signaling mechanism of Nox inhibitor-induced apoptosis was evaluated progressively using the verification of Bcl-2 family variation and inhibitor assay.
In this study, Nox2 inhibitor, GSK2795039, induced down-regulation of Mcl-1 and up-regulation of Bim and Noxa in a dose-dependent manner (as shown in Fig. 5). GSK2795039 induced the translocation of Bax and release of Cyt c. Previous studies reported that the translocation and conformational change of Bax leads to formation of outer-mitochondrial membrane spanning pores [26], and the Bax pores induce the release of Cyt c into the cytoplasm, which triggers activation of the caspase cascade [27]. These previous studies were also consistent with the findings in this study. Based on the current results, we propose an intra-cellular signaling mechanism by the Nox2 inhibitor, GSK2795039, as summarized in Fig. 8. The increase of Nox2 expression effectively causes ROS generation as an initial response in Raji cells. Both the Nox inhibitor GSK2795039 and ROS scavenger NAC effectively inhibit the cellular response following ROS generation. Aberrant ROS-mediated Bim and Noxa activation and Mcl-1 down-regulation induce the translocation of Bax to the mitochondria membrane, resulting in mitochondrial membrane pores. These Bax pores cause the release of Cyt c into the cytoplasm, inducing caspase cascade activation and apoptosis.
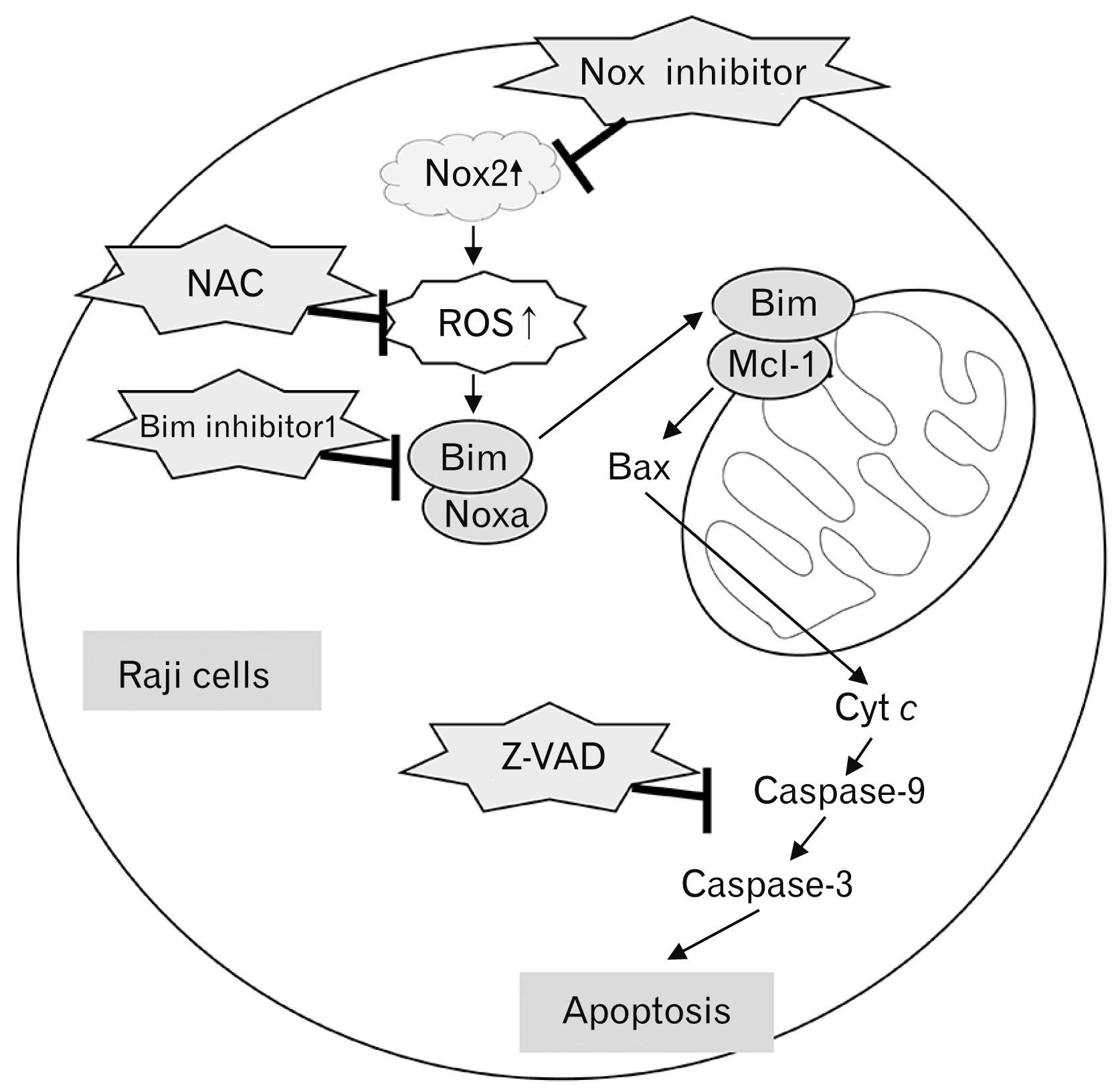 | Fig. 8Schematic diagram of the intracellular signaling mechanism during Nox2 inhibitor-induced apoptosis in Raji cells. The increase of Nox2 expression effectively causes ROS generation as an initial response in EBV-positive B lymphoma Raji cells. Both the Nox inhibitor and ROS scavenger NAC effectively inhibit the cellular response following ROS generation. Aberrant ROS-mediated Bim and Noxa activation and Mcl-1 down-regulation induce the translocation of Bax to the mitochondria membrane, resulting in mitochondrial membrane potential disruption. The Bax pores cause the release of Cyt c into the cytoplasm, resulting in caspase cascade activation and apoptosis. Cyt c, cytochrome c; EBV, Epstein-Barr virus; NAC, N-acetyl-L-cysteine; Nox2, nicotinamide adenine dinucleotide phosphate oxidase 2; ROS, reactive oxygen species.
|
In summary, Nox enzymes were highly expressed in EBV-positive Raji cells. The Nox specific inhibitor induced up-regulation of Bim and Noxa, down-regulation of Mcl-1, translocation of Bax, release of Cyt c, and caspase cascade activation in Raji cells, resulting in apoptosis. The precise molecular mechanism and action sequence of the Nox inhibitor is not yet clear and should be examined in further studies.
Go to :
 XML Download
XML Download