

 PDF
PDF Citation
Citation Print
Print



Introduction
Alzheimer’s disease (AD) and vascular dementia (VD) are the 2 most common subtypes of dementia [1]. AD is characterized by the progressive onset of impairments in memory- and execution-related performances, with concomitant cognitive problems such as aphasia, apraxia, and agnosia [2]. In contrast, VD is generally characterized by an acute onset with a time-dependent decline in memory and cognitive functions, combined with the apparent evidence of a cerebrovascular disease [3]. Therefore, accurate diagnoses should be the first step to cope with AD and VD [4]. However, the differential diagnosis still remains difficult, thus, the development of pharmacologic candidates can act on both AD and VD has rather received scientific attention in recent years [5,6].
There is increasing evidence of an association between AD and VD, possibly as a consequence of the vascular risk factors that are common to both pathologies [7]. Therefore, it might be beneficial to discover a common agent that can attenuate the hypoxic neuronal damage [8] that precedes these diseases. To elaborate, hypoxic neuronal damage commonly culminates in neuronal death by a pathologic cascade called “excitotoxicity” [9]. The diminished oxygen and glucose supplements in hypoxic conditions elicit an increment in glutamate release from the axon terminals of presynaptic neurons, which in turn causes excessive calcium entry into the dendritic spines of postsynaptic neurons. Subsequently, calcium triggers the overproduction of reactive oxygen species (ROS) by affecting mitochondrial dysfunction [10]. Since excitotoxicity appears to be inevitably linked with the accumulation of ROS, antioxidant treatments may be an appropriate strategy for coping with both AD and VD [11].
Considering the role of ROS in the development of hypoxic neuronal damage, numerous studies used experimental models of AD or VD [12,13] to screen for the possible effects of natural compounds, which are commonly known to possess ROS scavenging effects. Of these, phenolic compounds in plant tissues have been investigated in the context of developing
functional foods, since these compounds are key protectors of host plant tissues against oxidative injury [14]. Among the natural plants, Aster ageratoides Turcz. is a traditional medicinal organism in Korea that is particularly rich in phenolic compounds [15]. A recent study used HPLC-MSn to demonstrate that its aerial parts are particularly rich in chlorogenic acid (CGA, a phenolic compound) and identified 33 different CGAs in the Aster ageratoides extract (AAE) [16]. Together with the well-established anti-inflammatory [17], antibacterial [18], anticarcinogenic [19], and antioxidant activities [20] of CGAs, caffeic acid and quinic acid, two main CGA metabolites, have also been found to protect cultured cortical neurons against excitotoxicity [21]. Since excitotoxicity is the primary cause of pathogenesis in both AD and VD, AAE, which even in its crude form is highly enriched with CGA, may provide protection against both diseases by attenuating excitotoxicity. However, to the best our knowledge, no study has reported the possible modulatory effects of AAE in in vitro or in vivo models of either AD or VD.
In this study, we first evaluated whether AAE can protect PC12 cells (from the rat pheochromocytoma cell line) against excitotoxicity evoked by glutamate treatment. After confirming the attenuation of excitotoxicity by AAE in vitro, we further evaluated its protective effects on both AD and VD models, which were established by scopolamine-induced amnesia and 2-vessel occlusion and hypovolemia (2VO/H) techniques, respectively. To assess the anti-AD and -VD effects, we quantified the scopolamine-induced amnestic behaviors and 2VO/H-induced histopathologic changes in the hippocampus, respectively.
Go to : 
Materials and Methods
Preparation of Aster ageratoides extract
The aerial part of Aster ageratoides was collected in the Eumseong province in Korea on 30 May 2018, and its identification was confirmed by comparing it with the specimen preserved at the Department of Herbal Crop Research, National Institute of Horticultural and Herbal Science (NIHHS) (voucher No. MPS004392). The dried aerial part was powdered and extracted with distilled water as a solvent at room temperature (22°C–24°C). The water was subsequently eliminated by a freeze-dry process.
Cell culture
PC12 cells were purchased from the Korean Cell Line Bank (Seoul, Korea) and were cultured in an RPMI 1640 medium supplemented with 10% heat-inactivated fetal bovine serum and 1% penicillin/streptomycin at 37°C in a humidified atmosphere of 5% CO2. The culture media and all adjuvant reagents were purchased from GE Healthcare Life Sciences (Pittsburgh, PA, USA). The cultures were regularly checked and divided every third day.
MTT assay
PC12 cells were seeded in a 96-well plate at a density of 1×104 cells/well and incubated for 24 hours at 37°C. To find the lethal dose 50 (LD50) of glutamate, which is a concentration that can trigger excitotoxicity in PC12 cells, the cells were first subjected to glutamate insults with varying concentrations (0–50 mM) for 24 hours at 37°C. The LD50 was determined to be 50 mM glutamate (see Results), and the cells were co-treated with different concentrations (0, 10, 25, and 50 µg/ml) of AAE and 50 mM glutamate and subsequently incubated for 24 h at 37°C. Cell viability was determined using the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT; Sigma-Aldrich, St. Louis, MO, USA) assay. The MTT solution was created by diluting MTT in phosphate buffered saline (PBS) at a final concentration of 0.5 mg/ml. The MTT solution was added to each well, and the plate was incubated for 4 hours at 37°C. After incubation, the culture medium was removed, and the resulting insoluble, dark blue formazan crystal was dissolved with 100 µl dimethyl sulfoxide (DMSO; Sigma-Aldrich). The absorbance was measured at 540 nm in a microplate reader (ELx800UV; BioTek Instruments, Winooski, VT, USA), and the data were expressed as the percentage of viable cells relative to untreated controls.
Animals
A total of 40 male Sprague Dawley rats (8 weeks, 200–250 g) were purchased from Samtako (Osan, Korea) and housed in an environmentally controlled room at a constant temperature (21°C–23°C) and relative humidity (40%–60%) under a 12 hours light/dark cycle. All the rats had free access to water and food. The experiments were conducted in accordance with the Guide for the Care and Use of Laboratory Animals (National Institutes of Health publication, 8th Edition, 2011) [22]. Animal experiments in this study were approved by the Institutional Animal Care and Use Committee (approved protocol No: P-19-15-A-01) of Konyang University (Daejeon, Korea).
Experimental design
All rats were randomly assigned to 3 groups and treated differently as follows: with distilled water (n=20), with 10 mg/kg AAE (n=10), and with 50 mg/kg AAE (n=10). The AAE was dissolved in water as a vehicle to obtain a final volume of 1 mL, administered intraorally once a day for 7 days. At the 7th day of treatment, the rats in each group were further randomly divided into 2 subgroups (n=10, n=5, and n=5 for groups treated with the vehicle, 10 mg/kg AAE, and 50 mg/kg AAE, respectively). Subsequently, the 2 subgroups underwent distinct procedures for different experimental paradigms. Paradigm #1 aimed to evaluate the potential anti-AD efficacy of AAE, where the rats were injected with 1 mg/kg scopolamine via an intraperitoneal (i.p.) route, 30 min after the last administration of either the vehicle or AAE. The vehicle-pretreated rats without scopolamine treatment were used as control. Paradigm #2 aimed to evaluate the potential anti-VD efficacy of AAE, where the rats underwent either the 2VO/H surgery or sham-operation, 30 min after the last administration of either the vehicle or AAE. The vehicle-pretreated, sham-operated rats were used as control. In summary, the rats were divided into 4 groups for each paradigm, as follows (n=5 per group): CTRL, the group of controls; VEH, the vehicle-pretreated group with subsequent applications of scopolamine or 2VO/H; AAE-L, the group pretreated with low doses (10 mg/kg) of AAE and with subsequent applications of scopolamine or 2VO/H; and AAE-H, the group pretreated with high doses (50 mg/kg) of AAE and with subsequent applications of scopolamine or 2VO/H. All the rats assigned to paradigm #1 sequentially underwent 3 different behavioral analyses (the Y-maze, novel object recognition, and passive avoidance tests) and were subsequently euthanized. In contrast, the rats assigned to paradigm #2 were further administered with the vehicle or AAE until postoperative day (POD) 7 and subsequently sacrificed for obtaining brain samples. The experimental designs are illustrated in Fig. 1A and B.
 | Fig. 1Timelines of the experimental protocols and time-dependent changes in rCBF values during the 2VO/H operation. (A) For experimental paradigm #1, 20 rats (n=5 per group) were pretreated with the vehicle or AAE for 7 days and subsequently treated with scopolamine (i.p.). Using these rats, 3 different behavioral tests were conducted. For experimental paradigm #2, rats (n=5 per group) with the same treatment schedule underwent the 2VO/H surgery, and their brains were obtained for histologic analyses at POD 7. (B) Detailed protocols of 3 behavioral tests. (C) The rCBF values throughout the 2VO/H operation, as detected by a laser-Doppler flowmetry. AAE, Aster ageratoides extract; C-V, cresyl violet; IHC, immunohistochemistry; NORT, novel object recognition test; PAT, passive avoidance test; POD, postoperative day; rCBF, relative cerebral blood flow; Y-MT, Y-maze test; 2VO/H, 2-vessel occlusion and hypovolemia.
|
Y-maze test
The Y-maze test (Y-MT) was employed as the first tool to assess memory performance for experimental paradigm #1. The scopolamine-treated rats were initially introduced to the center of the matte black plastic maze, with 3 arms, each of length 50 cm, width 15 cm, and height 30 cm, at 120-degree intervals. The sequence and number of arm entries were monitored for an 8-minutes period. To calculate the percentage of spontaneous alternation behavior, the following formula was used: Spontaneous alternation (%)=[(Number of alternations)/(Total number of arm entries-2)]×100. The number of total arm entries also served as an indicator of locomotor activity. After each rat was tested, the maze was cleaned using 70% ethanol.
Novel object recognition test
The novel object recognition test (NORT) was employed as the second tool to assess memory performance for experimental paradigm #1. Twenty-four hours after the Y-MT, the rats were given the different treatments and scopolamine according to the same protocols used in the Y-MT and underwent the familiarization session of NORT. Subsequently, they were introduced to the center of the 70×70×35-cm-sized matte black square box containing 2 identical objects spaced diagonally at 30-cm intervals. The rats were allowed to freely explore the objects for 10 minutes. After 24 hours, the rats underwent the test session, at which point one of the familiar objects was replaced by a novel object. The rats were again allowed to freely explore the objects for 10 minutes. The positivity of object preference was judged by a rat directing its nose to within 2 cm of the object, touching it, rearing on it, or staying beside it for over 5 s. The exploratory behavior of individual rats during the familiarization and test sessions was recorded and analyzed with the aid of a video camera connected to an EthoVision XT9 system (Noldus, Wageningen, the Netherlands). After each rat was tested, the box and the objects were cleaned using 70% ethanol.
Passive avoidance test
The passive avoidance test (PAT) was employed as the third tool to assess the memory performance for experimental paradigm #1. The PAT apparatus was equipped with 2 juxtaposed illuminated and dark chambers, each 25×20×25 cm in size. A 50-W lamp was placed 1 m above 1 chamber for illumination. Each test involved 2 separate sessions: a training session and a test session. Prior to the training session, the rats were administered the different treatments and scopolamine according to the same protocols used in the previous tests. During the training session, the rats were initially placed in the illuminated chamber and, on entering the dark chamber, received an electrical shock (0.5 mA, delivered through stainless steel rods) for 3 s. Once the rats had entered the dark compartment, the “initial” latency periods were measured using a stopwatch. A test session was performed 24 hours after the training session, and the “step-through” latency times to re-enter the dark chamber were measured up to 100 s. After each rat was tested, the walls of the chambers were cleaned using 70% ethanol.
2VO/H
2VO/H was employed to establish an animal model of VD using a modified version of a previously described method by Smith et al. [23]. For this, the rats were anesthetized with 5% isoflurane in 70% N2O and 30% O2 and maintained during the operation at a level of 1.5%–2% isoflurane under spontaneous respiration. Throughout the operation, the rectal temperature was controlled at 37°C with a heating pad. The left femoral artery was exposed and catheterized with a PE-50 catheter to allow the future withdrawal of blood to cause hypovolemia. The right jugular vein was isolated and injected with 500 units/kg heparin dissolved to 100 units/ml with 0.9% saline. The bilateral common carotid arteries (CCAs) were exposed and permanently ligated with a 4-0 nylon suture. Following this, blood was withdrawn from the femoral artery via a catheter to reduce cerebral blood flow (CBF). When the relative CBF (rCBF) value, as measured by Laser-Doppler flowmetry (Periflux 5000; Perimed AB, Järfälla-Stockholm, Sweden), reached below approximately 20% of the baseline, the blood withdrawal was stopped for 15 minutes to maintain the ischemic period. During this period, a syringe filled with the withdrawn blood was kept in shaking water bath, maintained at 37°C to prevent coagulation. The blood was subsequently reinfused, and the rCBF rapidly returned to nearly 60% of the baseline. After the rCBF value plateaued, the animals regained consciousness and were maintained in their home cage until POD 7. The representative Laser-Doppler flowmetry for tracing rCBF values during this procedure is presented in Fig. 1C.
Cresyl violet staining
On POD 7, the rats were transcardially perfused with 4% paraformaldehyde (PFA). Their brains were isolated, post-fixed with 4% PFA, dehydrated with a graded ethanol series, embedded in paraffin, and serially sectioned at 5-µm thickness using a microtome (RM2255, Leica, Nussloch, Germany). Two slides were randomly selected from the hippocampus-bearing tissue collections of each rat, deparaffinized in xylene, hydrated by a decreasing ethanol gradient series, and washed twice in distilled water. The slides were subsequently stained with a 0.1% cresyl violet solution (Sigma-Aldrich). The hippocampal CA1 regions were photographed at 200x magnification by a digital camera connected to a light microscope (DM4, Leica). From the images, the number of intact neurons, which included those with clear nuclei and large cell bodies, in the CA1 subfield (localized within 300 μm) were counted and averaged.
Immunohistochemistry
For immunohistochemistry (IHC), the 2 washed slides from each rat were incubated with rabbit anti-glial fibrillary acidic protein (GFAP; Zymed Laboratories, San Francisco, CA, USA) and rabbit anti-Iba-1 (Wako, Richmond, VA, USA), each diluted in PBS to a ratio of 1:200, in a humid chamber for 24 hours at 4°C. After 3 washes in PBS, each slice was incubated with biotinylated anti-rabbit IgG antibodies (Vector laboratories, Burlingame, CA, USA), which were diluted in PBS to a ratio of 1:250, in a humid chamber for 2 hours at 22°C–24°C. After 3 washes in PBS, the slides were further incubated with an avidin-biotin complex (VECTASTAIN ABC kit; Vector laboratories), which was diluted in PBS to a ratio of 1:250, for 1 hours at 22°C–24°C. The resulting immunoreactivities turned to a dark brown color after the addition of the chromogen, 3,3'diaminobenzidine (DAB; Vector laboratories). After mounting, the hippocampal CA1 regions of each slide were photographed at 200x magnification using a digital camera connected to a DM4 light microscope (Leica). In each photograph, the number of cells (localized within 300 μm) with either Iba-1 immunopositive reactions or GFAP-immunoreactivity were quantified using Image J (v1.49, National Institutes of Health) [24].
Statistical analyses
All data are presented as the mean±standard error of the mean (SEM). The inter-group comparisons were conducted using one-way analysis of variance (ANOVA) in GraphPad Prism 5 (GraphPad Sotfware Inc., San Diego, CA, USA). P<0.05 was considered to indicate a statistically significant difference.
Go to :
Results
AAE attenuates neuronal excitotoxicity in vitro
As described above, excitotoxicity is a major factor in the pathogenesis of both AD and VD. Therefore, AAE was first tested for its potential protective effects against excitotoxic neuronal death using PC12 cells co-treated with glutamate. Considering that the approximate LD50 value of 24-hours glutamate treatment in this cell line was 50 mM (arrowhead; Fig. 2A) and that the cell viabilities were unaffected by AAE concentrations below 50 µg/ml (data not shown), we next evaluated the cell viabilities after 24 hours of co-treatment with 50 mM glutamate and different concentrations of AAE (0, 10, 25, and 50 µg/ml; Fig. 2B). The results demonstrated that glutamate treatment triggered the apparent excitotoxicity in PC12 cells (***P<0.001 vs. control), which was attenuated by co-treatments with AAE in a dose-dependent manner (###P<0.001 vs. the cells treated with glutamate alone; δδδP<0.001 vs. the cells co-treated with glutamate and 10 µg/ml AAE), indicating that AAE can attenuate in vitro neuronal death triggered by excitotoxicity.
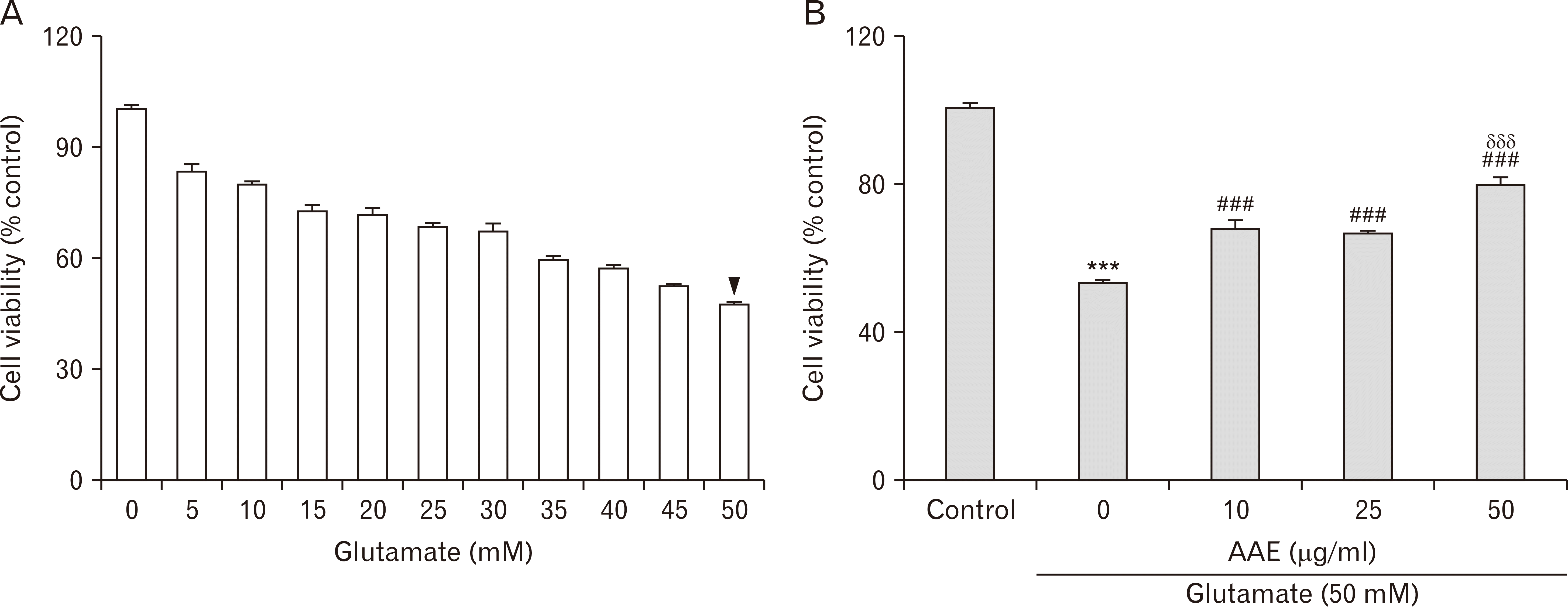 | Fig. 2The effects of AAE on PC12 cell viability in vitro, following glutamate-induced excitotoxicity. (A) Dose-dependent cell viability following a 24 hours challenge with varying concentrations of glutamate. The approximate LD50 value for this cell line after 24 hours of glutamate insult was 50 mM (arrowhead). (B) The effects of 24 hours of co-treatment with AAE (0, 10, 25, and 50 µg/ml) and glutamate (50 mM) on PC12 cell viability. Values are presented as the mean±standard error of the mean (***P<0.001 vs. control; ###P<0.001 vs. the cells treated with only glutamate; δδδP<0.001 vs. the cells co-treated with glutamate and 10 µg/ml AAE). AAE, Aster ageratoides extract; LD50, lethal dose 50.
|
AAE attenuates memory impairment in vivo in an AD rat model
In experimental paradigm #1, we investigated whether AAE can prevent memory impairment, which is an essential feature in AD patients. For this, we assessed the effects of AAE intake on AD-associated memory impairment in a rat model with scopolamine-induced amnesia [25] and subsequently applied 3 different behavioral analyses, i.e., Y-MT, NORT, and PAT, on the rats. The Y-MT results indicated that the VEH group experienced significant impairments in memory function, as indicated by the changes in spontaneous alternation behavior (***P<0.001 vs. CTRL; Fig. 3A). However, the AAE-H group showed a significant increase in spontaneous alternation behavior compared with the VEH group (#P<0.05 vs. VEH; Fig. 3A), while the AAE-L group showed no such change. Neither scopolamine nor AAE affected the locomotor activity significantly, as measured by the number of total arm entries (Fig. 3B). The NORT results demonstrated that all groups spent similar amounts of time exploring 2 identical objects (Fig. 3C) during the familiarization session. In the test session, while the CTRL group spent significantly more time exploring the new object than the familiar one (*P<0.05; Fig. 3D), the VEH and AAE-L groups lacked this tendency. However, the AAE-H group explored the new object for a significantly longer time than the familiar one (***P<0.001), suggesting that the scopolamine-induced impairment of memory performance was reversed by the oral administration of AAE (50 mg/kg or higher). Neither scopolamine nor AAE affected locomotor activity significantly, as shown by the total distance moved (Fig. 3E). Finally, the effect of AAE on AD-associated memory impairment was also validated by the PAT results, which showed that the VEH group developed a significant impairment in retention memory compared to the CTRL group, as shown by lower step-through latencies (***P<0.001 vs. CTRL; Fig. 3F) in test sessions. However, the step-through latencies of the AAE-L and AAE-H groups were significantly longer than that of the VEH group (#P<0.05 and ###P<0.001, respectively), and these changes were dose-dependent (δP<0.05 vs. AAE-L, Fig. 3F). Neither scopolamine nor AAE affected the initial latencies assessed during training sessions (Fig. 3G). Taken together, these results suggested that the oral administration of AAE can attenuate memory impairment in a rat model of AD.
 | Fig. 3The effects of AAE on memory-related performances in rats with scopolamine-induced amnesia (n=5 per group). The average spontaneous alternation rate (A) and the number of total arm entries for >8 minutes (B) during the Y-maze test. The percentage of time spent exploring the 2 familiar objects during the familiarization session (C), the novel object over the familiar object during the test session (D), and the total distance moved (E) during the novel object recognition test. The step-through latencies (F) and the initial latencies (G) to enter the dark chamber during the test and training sessions, respectively, during the passive avoidance test. Values are presented as the mean±standard error of the mean (***P<0.001 vs. CTRL; #P<0.05 and ###P<0.001 vs. VEH; δP<0.05 vs. AAE-L). AAE, Aster ageratoides extract; AAE-H, high dose of AAE-treated group; AAE-L, low dose of AAE-treated group; CTRL, control group; N.S., statistically not significant; VEH, vehicle-treated group.
|
AAE attenuates deteriorations in hippocampal structures in vivo in a VD rat model
In experimental paradigm #2, we evaluated the anti-VD efficacy of AAE by using the 2VO/H technique, a surgical model that mimics clinical VD [26], by investigating whether AAE intake can prevent morphological deterioration in the hippocampal structure (an essential feature in both 2VO/H-induced VD rats and VD patients). For this, we used cresyl violet staining to assess the effects of AAE pretreatment (7 days) on the 2VO/H-induced loss of pyramidal neurons in the hippocampal CA1 region. At POD 7, the VEH group showed a dramatic decrease in the number of hippocampal CA1 neurons compared to that of the CTRL group (***P<0.001 vs. CTRL; Fig. 4A and B). The VEH group also showed the presence of apoptotic neurons, which were characterized by pyknotic nuclei and shrunken cytoplasm, in the stratum pyramidale (SP) of the hippocampal CA1 region (arrows in Fig. 4A, VEH). However, the number of surviving neurons was significantly higher in both the AAE-L and AAE-H groups (##P<0.01 and ###P<0.001 vs. VEH), and this change was dose-dependent (δδδP<0.001 vs. AAE-L). Since neuroinflammation is commonly associated with neuronal death [27], we next quantified the extent of microgliosis and astrocytosis in the same region using immunohistochemical detection methods (Iba-1 and GFAP antibodies, respectively). The VEH group had a significantly higher number of Iba-1 immunopositive (+) microglia in the SP, stratum oriens, and stratum radiatum compared to the CTRL group (Fig. 4C). The number of activated microglia in the hippocampal CA1 of the VEH group were approximately 10 times higher than that of the CTRL group (**P<0.01; Fig. 4D). However, the number of activated microglia were significantly lower in the AAE-L group (#P<0.05 vs. VEH), and this reduction was more exaggerated in the AAE-H group (##P<0.01 vs. VEH; δP<0.05 vs. AAE-L). Contrary to expectations, there were no inter-group differences in the optical density of GFAP+ astrocytes (Fig. 4E and F). Collectively, these results suggested that oral administration of AAE could prevent both the VD-associated neuronal loss and the associated microglial activation.
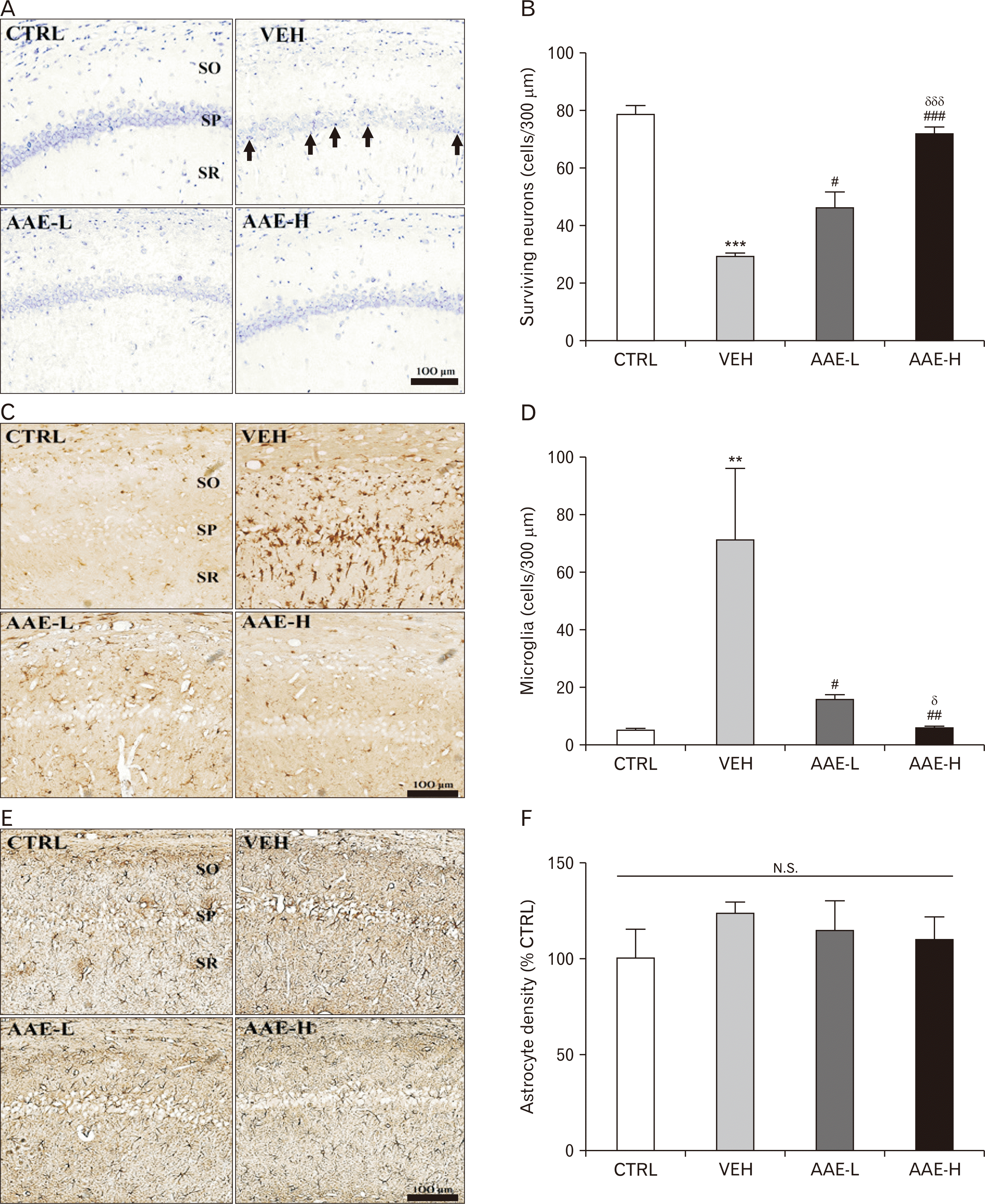 | Fig. 4The effects of AAE on the number of surviving neurons, and the extent of neuroinflammation in the hippocampal CA1 of rats (n=5 per group) at POD 7. A representative cresyl violet-stained image (A) and the graph showing the average number of surviving neurons (B) in the hippocampal CA1. A representative immunohistochemistry image of the hippocampal CA1 using anti-Iba-1 (C) and anti-GFAP (E) and the quantitative graphs (D and F, respectively). The region of interest was within 300 µm in width in the hippocampal CA1 region. Values are presented as the mean±standard error of the mean (**P<0.01 and ***P<0.001 vs. CTRL; #P<0.05 and ###P<0.001 vs. VEH; δP<0.05 and δδδP<0.001 vs. AAE-L). AAE, Aster ageratoides extract; AAE-H, high dose of AAE-treated group; AAE-L, low dose of AAE-treated group; CTRL, control group; GFAP, glial fibrillary acidic protein; N.S., statistically not significant; POD, postoperative day; SO, stratum oriens; SP, stratum pyramidale; SR, stratum radiatum; VEH, vehicle-treated group.
|
Go to :
Discussion
Increasing knowledge of the cellular events underlying hypoxic neuronal damage, such as excitotoxicity, has inspired researchers to screen compounds capable of ceasing or, at least, decelerating the progress of neurodegenerative such as AD and VD [28]. In hypoxic conditions, the diminished levels of oxygen and glucose supplementation trigger the depolarization of presynaptic neurons and excessive release of the neurotransmitter glutamate into the synaptic cleft. Excessive glutamate subsequently causes prolonged stimulation of N-Methyl-D-aspartate (NMDA) receptors in the dendritic spines of postsynaptic neurons and can also increase calcium influx into the neuron [29]. The entry of calcium through NMDA receptors can be devastating because of at least 2 reasons: 1) cellular attempts to get rid of the excess calcium use up already scarce supplies of ATP and 2) excessive influx of calcium causes the disordered activation of a wide range of enzyme systems such as proteases, lipases, and nucleases, which in turn disassemble cell membranes, genetic material, and structural proteins in the neurons, ultimately leading to cell death [28,30]. Indeed, neuronal excitotoxicity is a quite complex process involving a series of molecular and cellular events as described above. Therefore, there exists an urgent need for research to develop therapeutic agents with multiple-target properties.
Natural compounds are complex multiple-target molecules, found mainly in plants. To date, these have been extensively studied for their antioxidant properties. However, they are known to modulate diverse signal transduction cascades through direct effects on enzymes including various kinases, regulatory proteins, and receptors [31,32]. Furthermore, some polyphenols also exert health-promoting effects through epigenetic modifications [33]. Due to their broad-spectrum effects, natural compounds are suitable candidates for the treatment of multifactorial diseases such as cancer [34] and neurodegenerative diseases [35] including dementia [36].
The present study demonstrated the protective potential of AAE on rats with scopolamine-induced amnesia and 2VO/H, which were used as experimental models for AD and VD, respectively. Considering the increasing evidence for the involvement of excitotoxicity in both pathologies [37], we used PC12 cells which has known to possess diverse glutamate receptors in their membrane, as a victim of excitotoxicity in this study. Using this method, we revealed that AAE protected PC12 cells against glutamate-induced excitotoxicity.
This study employed and 2 distinct in vivo models, scopolamine-induced amnesia and 2VO/H as screening platforms to assess the therapeutic effects of AAE for AD and VD, respectively. Scopolamine-induced amnesia has been used extensively to screen the effects of diverse compounds with various origins and formats for development as therapeutic agents for AD. In fact, the memory impairment produced in rats following intraperitoneal injections of scopolamine resembles the clinical symptoms of AD patients [38]. Scopolamine influences the expression of several genes associated with muscarinic receptor signaling pathways, apoptosis, and cell differentiation in the rat brain [39]. The 2VO/H technique aimed to model global cerebral ischemia, which occurs in VD patients and essentially comprises both permanent bilateral CCA occlusion and hypovolemic conditioning. This model produces pathophysiologic changes including ischemic neuronal damage, neuroinflammatory changes in the hippocampus, and the impairment of hippocampus-related behaviors such as cognition, spatial learning, and memory [40].
The in vivo results in this study indicated that AAE treatments can attenuate scopolamine-induced memory impairment, as well as 2VO/H-induced neuronal death and microglial activation in the hippocampus. The GFAP+ reaction (a surrogate of astrocyte activation [41]) in the hippocampal CA1 region was also quantified in this study; however, it failed to detect any inter-group differences. One plausible explanation involves the timing of observations in hippocampal CA1 tissues undergoing pathologic changes after ischemic insults. In fact, in diseases such as Parkinson’s disease, AD, multiple sclerosis, and human immunodeficiency virus-induced dementia, microglial activation commonly precedes astrogliosis and overt neuronal loss [42]. There is increasing evidence to suggest that activated microglia are essential for the development of activated astrocytes as they release soluble factors that stimulate signaling pathways in astrocytes, leading to the production of various inflammatory mediators [43].
This study provided evidence that AAE supplements can exert anti-AD- and -VD effects, suggesting that AAE might be an edible phytotherapeutic candidate for the 2 major types of dementia, exerting the cognition-promoting and neuroprotective effect. The major limitation of this study is the lack of proposed detailed mechanisms underlying the AAE-mediated inhibition of excitotoxicity in both in vitro and in vivo conditions. Excitotoxicity and the associated dementia involve complex pathological cascades including a series of molecular and cellular events such as apoptosis [44] and mitochondrial dysfunction [45]. Therefore, more advanced and well-designed studies are needed in the future, so as to provide a more detailed explanation of the specific mechanisms underlying AAE-mediated protection against dementia.
Go to :
 XML Download
XML Download