

 PDF
PDF Citation
Citation Print
Print


Trisomy 9 mosaicism is a rare chromosomal abnormality. Since its first mention in 1973 by Haslam et al.,1 approximately 150 cases have been reported worldwide.2 In Korea, Kim et al. described a case of trisomy 9 mosaicism, which had been mistaken as Smith–Lemli–Opitz syndrome.3
Trisomy 9 mosaicism is characterized by intrauterine growth retardation (IUGR), mental retardation, craniofacial deformities, skeletal abnormalities, congenital heart defects, and genital abnormalities. The incidence and severity of the accompanying malformations and mental retardation correlate with the percentage of trisomic cells.4,5
Here, we report a case of a neonate with trisomy 9 mosaicism that, was confirmed by single nucleotide polymorphism array and fluorescence in situ hybridization tests, even though the results of the initial peripheral blood genetic testing were normal.
CASE
The male infant was delivered by emergent cesarean section at 36 and 5/7 weeks of gestation. The patient was the first child of non-consanguineous parents. The mother of the patient was 32 years old and denied that she had any exposure to radiation or medication during pregnancy. She was diagnosed with hypothyroidism 2 years earlier, but no medication was administered. Furthermore, she had no specific family history associated with any hereditary disorder or chromosomal abnormalities. The mother had undergone regular check-ups at a local gynecology clinic. During the routine prenatal examination at the gynecology clinic, UGR with oligohydramnios was observed beginning in the third trimester; thus, she was referred to our hospital. However, on arrival at our out-patient clinic, we detected fetal heart rate deceleration ion and poor variability. Therefore, emergent caesarian section was indicated. A prenatal cytogenetic study was not performed.
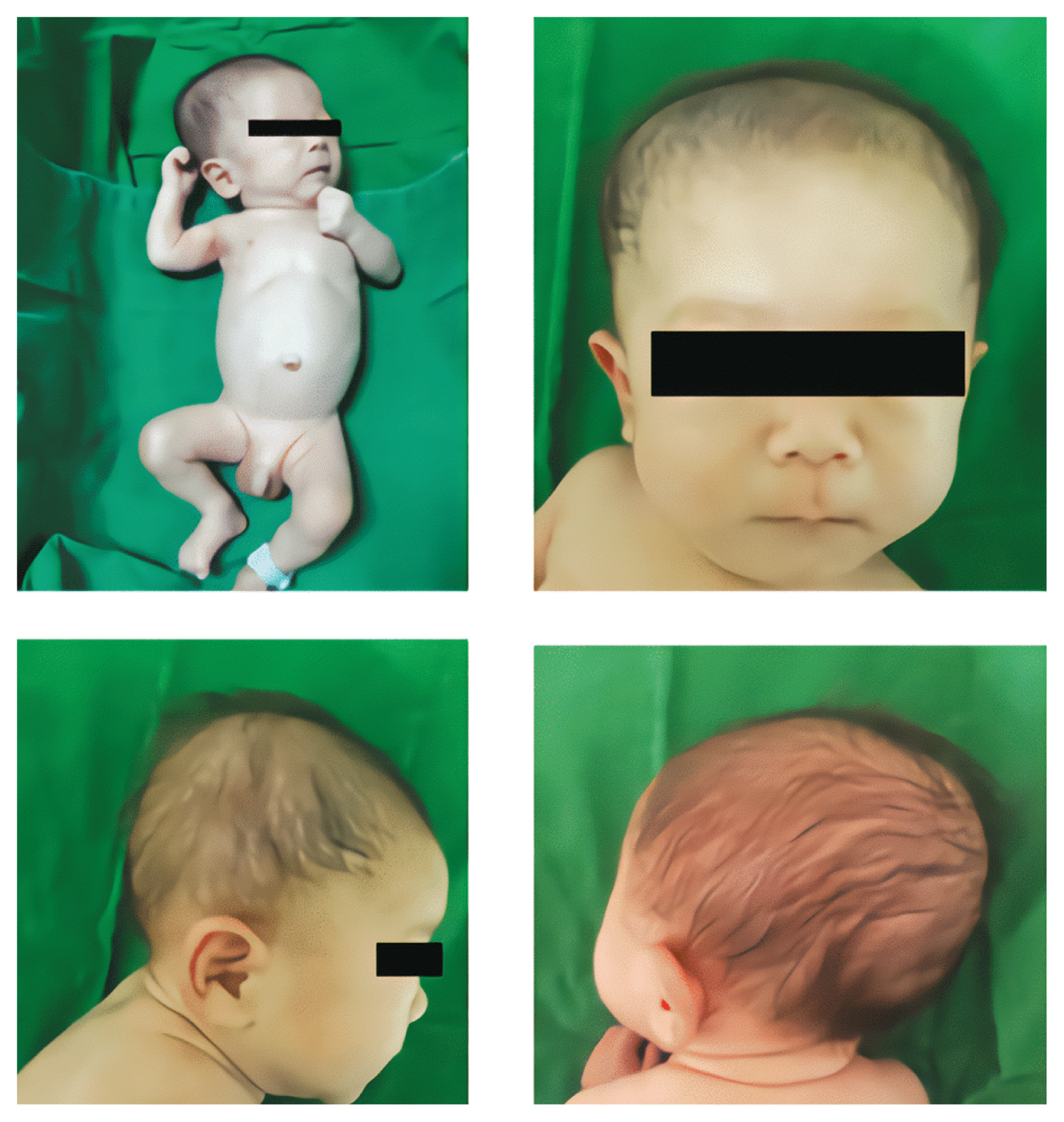
The weight of the neonate was 1,350 g, length was 41.0 cm, and head circumference was 29.5 cm, all of which were less than the 10th percentile. The patient was admitted to our neonatal intensive care unit shortly after birth due to respiratory difficulties. Upon presentation, the patient had a dysmorphic face with a broad nasal root; small fish-shaped mouth with thin lower lips; retro-micrognathia; low-set, malformed and posteriorly rotated ears; microcephaly; and plagiocephaly (Fig. 1). Physical examination revealed a grade II systolic murmur at the left suprasternal notch.
The initial chest radiograph showed decreased lung volume with air bronchogram. The patient was initially treated with surfactant following the INSURE technique, and then, a nasal positive pressure ventilator was applied for respiratory support. Results of the routine laboratory test were as follows: white blood cell count, 4,400/mm3; hemoglobin level, 16.7 g/dL; platelet count, 99,000/mm3; Na+/K+/Cl− concentrations, 139/4.5/105 mEq/L; total protein/albumin level, 4.9/3.3 mg/dL; BUN/Cr concentration, 11.7/0.85 mg/dL; aspartate aminotransferase/alanine aminotransferase level, 48/5 U/L; and C-reactive protein level, 0.01 mg/dL. Complete blood tests showed no abnormalities other than mild thrombocytopenia. Several tests were performed to assess for the presence of metabolic disorders and congenital infection. Metabolic evaluation showed normal results. Moreover, the patient tested negative for herpes simplex, cytomegalovirus, rubella, and toxoplasma. Initial brain sonography revealed increased periventricular echogenicity. The abdominal ultrasonographic examination showed no abnormal findings.
On the 2nd day after birth, enteral feeding was initiated, through an orogastric tube and was well tolerated. Full enteral nutrition was achieved by on the 16th day after birth.
On the 3rd day after birth, the patient’s echocardiogram showed perimembranous ventricular septal defect (VSD) measuring 4.0 mm, atrial septal defect (ASD) measuring 3.5 mm, and a small patent ductus arteriosus. The follow-up echocardiogram performed on the 17th day after birth revealed a closed ductus arteriosus with the VSD measuring 4.0 mm and the ASD measuring 2.5 mm.
On the 6th day after birth, the patient’s platelet count decreased to 6,000/mm3; hence, intravenous immunoglobulin (IVIG) was administered (1 g/kg/d1 for 2 days) under suspicion of neonatal allo-immune thrombocytopenia because mother’s platelet count was normal (284,000/mm3). Shortly after the initiation of IVIG treatment, the patient’s platelet count increased to 38,000/mm3 and then gradually returned to a normal level 20 days after performing this series of laboratory tests. Since then, his platelet count has remained normal.
On the 13th day after birth, the direct bilirubin level of the patient increased to 1.64 mg/dL, and his total bilirubin was 8.64 mg/dL. Work-ups for cholestasis including abdominal sonography, viral marker and other blood tests (TORCH and thyroid function test) all showed negative findings; hence, the patient was treated under the assumption of parenteral nutrition – associated cholestasis (PNAC). Therefore, we applied a more aggressive enteral nutritional therapy including a rapid increase in the amount of enteral feeding, a limitation on the maximum intake of parenteral amino acid solution, and removal of copper and manganese from the parenteral nutrition solution in combination with ursodeoxycholic acid (UDCA) treatment. On the 62nd day after birth, direct bilirubin had normalized and UDCA treatment was discontinued.
Brain magnetic resonance imaging performed on the 63rd day after birth showed on abnormal findings.
After gradual weaning off the ventilator, respiratory support was discontinued on the 13th day after birth. On the 15th day after birth, bottle feeding was started; however, it took a while for the patient to tolerate bottle feeding due to feeding hypoxia. Finally, full bottle feeding was achieved on the 60th day after birth. On the 77th day after birth, the patient was discharged from the hospital with the following assessment findings: weight, 2,850 g (less than the 10th percentile); height, 46 cm (less than the 10th percentile); and head circumference, 36 cm (10–25th percentile).
Genetic testing
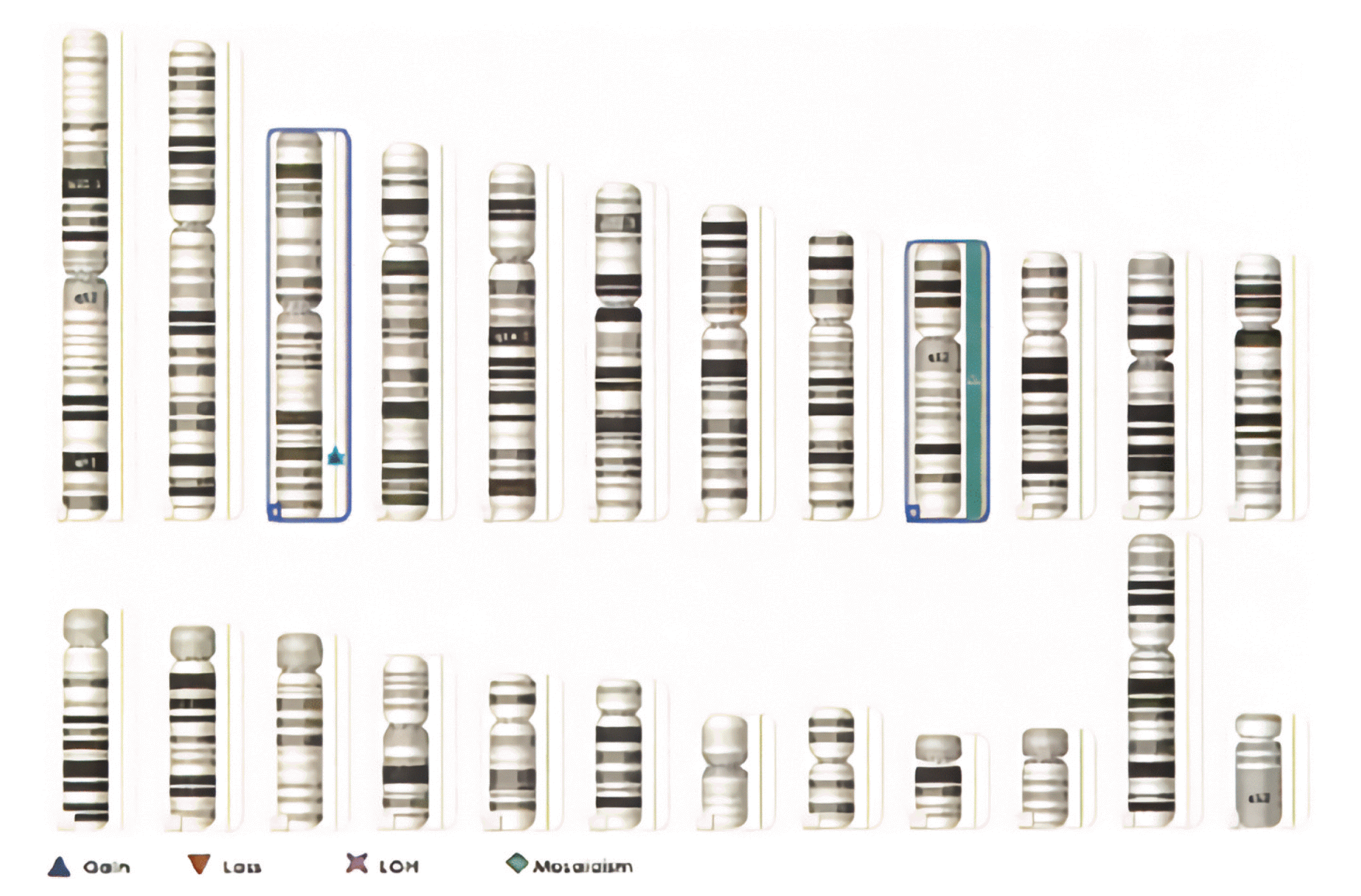
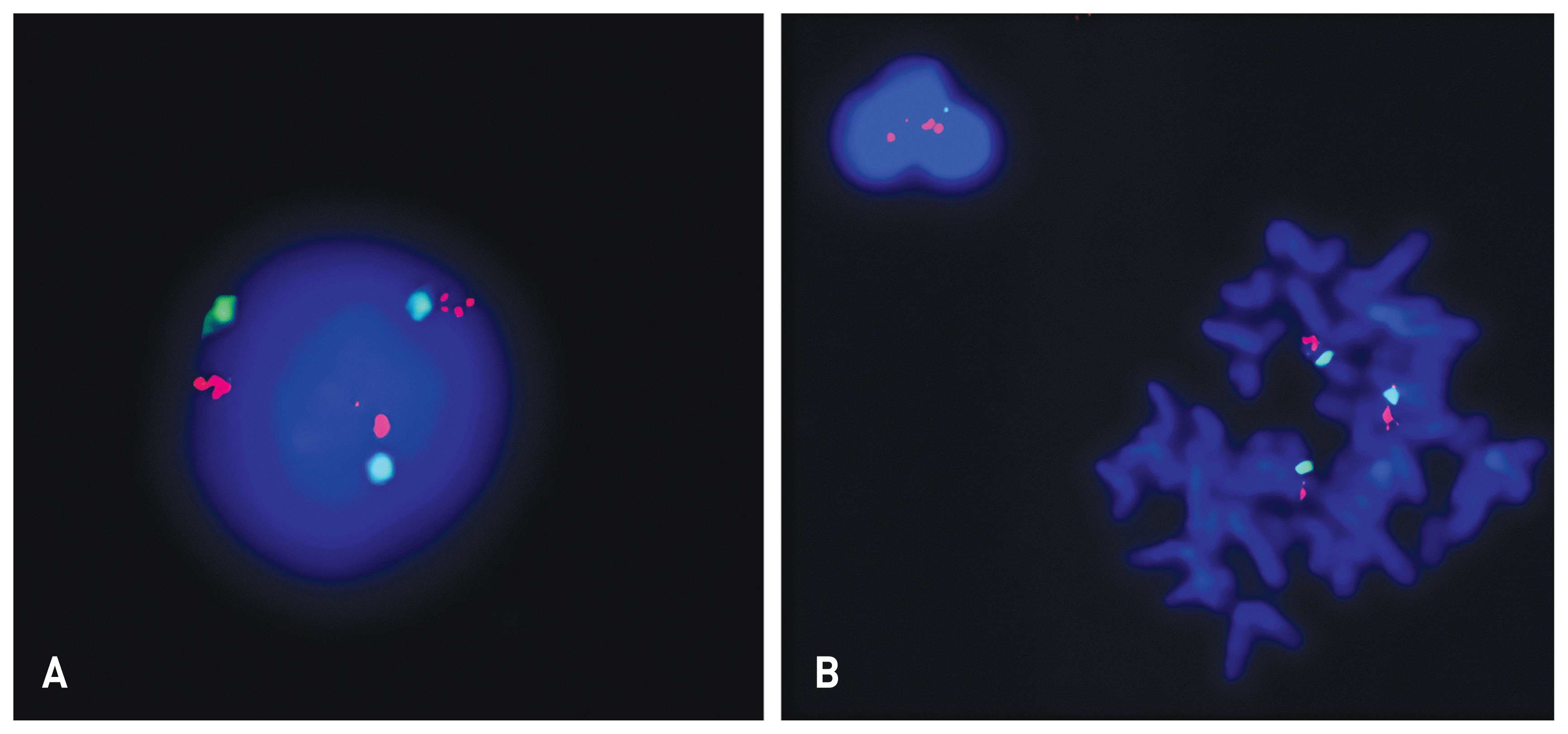
Chromosome tests were performed on peripheral blood lymphocytes based on the suspicion of chromosomal abnormalities due to the presence of IUGR, facial dysmorphism, and cardiac defects. A balanced translocation was found from the analysis of 20 dividing cells obtained by culturing from peripheral blood (46, XY, t(2;9)(p22;q33)). Microarray analysis showed a trisomy 9 mosaicism. Microarray analysis was performed using the CytoScan 750K high-resolution single nucleotide polymorphism (SNP) array (Affymetrix, Santa Clara, CA). The array contains > 750,436 copy number variant markers, including 200,436 genotype-able SNP probes and 550,000 non-polymorphism probes. All data were visualized and analyzed using the Chromosome Analysis Suite software package (Affymetrix, USA). The analysis showed a translocation 46,XY,t(2;9)(p22;q39). Based on a comparison with human reference genome 37 (NCBI37/hg19) at the National Center for Biotechnology, a trisomy 9 mosaicism and a 567 Kb duplication which was located in 3q26.1 [arr 3q26.1 (164876489_165443782)x3] were identified (Fig. 2). The fluorescence in situ hybridization (FISH) test showed 10–15% of trisomy 9 cells among 200 cells (Fig. 3).
Outcome
The patient is doing relatively well after discharge. On the last outpatient clinic follow up, the patient was 8 months of age. He was feeding with a bottle and had recently started a soft diet. The VSD had reduced to 1mm in size and there were no symptoms of heart failure, therefore the defect is being observed without surgery. Development of the patient was suitable for a normal 6 to 7 old infant, such as making continuous, vowel and, consonant sounds, and responding to his name. Additionally, he could roll supine to prone and sit down with minimal support. His body measurements were as follow; body weight 6.5kg (< 10th percentile), height 67cm (< 10th percentile), and head circumference 42.0cm (10–25th percentile).
Go to : 
DISCUSSION
A total of 150 cases of chromosome 9 trisomy mosaicism have been reported since it was first reported in 1970 by Haslam et al.1 In Korea, Jeon et al. reported the first case of trisomy 9 syndrome in 1998.6 Kim et al. described the first case of trisomy 9 mosaicism, which was mistaken as Smith–Lemli–Opitz syndrome in 2001.3 Since then, only 3 additional cases have been reported: Lee et al.,7 Kim et al.,8 and Na et al.9 in 2002, 2003, and 2016, respectively. Trisomy 9 is subdivided into two types: the complete type, with one additional complete chromosome 9; and the partial type, with a short arm or a part of the long arm of chromosome 9. The partial type is more common than the complete type; in Korea, cases of partial trisomy 9 have been reported in 199510 and 200011
The complete type is further subdivided into mosaic type and non-mosaic type. Most of the fetuses with non-mosaic type have been aborted or died shortly after birth.4 The average survival time of neonates with non-mosaic type is less than 9 days. The mosaic type is more common than the non-mosaic type and has a better survival rate. It has been reported that some infants with low-level mosaic type can survive for more than 2 years.12
The clinical features of infants with trisomy 9 mosaicism are diverse, and, less severe than those of complete types. Infants with partial trisomy 9 may survive until adulthood. The degree of malformation is more severe according to the proportion of trisomy 9 cells.4,5 Clinical manifestations include microcephaly, micrognathia, narrow temples, dysmorphic face including protruding larynx, thick and wide nose, low-set ears, and fish-shaped mouth. It may be accompanied by genitourinary malformations and central nervous system malformations.12
The complete type has a higher rate of natural abortion and postpartum mortality than the partial type; hence, prenatal diagnosis is very important. 13 Ultrasound is a very useful method for assessing trisomy 9 mosaicism during the prenatal period. Fetal umbilical cord blood or amniotic cytogenetic testing can be performed if trisomy 9 mosaicism is suspected based on the ultrasound results, but the diagnosis rate is low. When the presence of trisomy 9 cells has been determined, FISH can accurately detect the presence of these cells in metaphase and interphase. In addition, cases of mosaicism have been reported after a repeat FISH of blood and skin fibroblasts was performed in patients diagnosed with non-mosaic type by conventional cytogenetic tests. The majority of the patients with longer than expected survival were diagnosed with mosaic type after performing a repeat FISH.14
Since our case showed shortness of breath and low birth weight, delayed intrauterine growth, and craniofacial dysmorphism, we suspected this patient may have chromosomal abnormalities. Thus, an initial peripheral blood chromosome test was performed. A balanced translocation was found from the analysis of 20 dividing cells obtained by culturing from peripheral blood (46, XY, t(2;9)(p22;q33)). Then, we performed an additional microarray analysis to find any microdeletions at the translocation site, but interestingly, the analysis revealed a trisomy 9 mosaicism. When we rechecked 50 dividing cells from peripheral blood after confirming trisomy 9 in the microarray test, we could not find any trisomic cells. An additional FISH test showed 10–15% of trisomy 9 cells among 200 cells. When the fraction of cells with abnormal karyotypes is low, there is a possibility that the abnormal cells will die before detection in cell culture during routine peripheral blood chromosome testing. Therefore, even if the result of the peripheral blood chromosome test is normal, it is recommended that additional microarray tests should be considered if the patient is suspected to have a chromosomal abnormality. The 4 previously reported cases of trisomy 9 mosaicism in Korea were all confirmed from initial peripheral blood karyotyping test. It is reasonable to consider the possibility that the proportion of abnormal cells in our patient was less than other patients, and that is also the reason for showing a better prognosis than others. The reason for this is that the proportion of cells with a normal karyotype is high and the degree of malformation is relatively mild; hence, patients with this condition may survive longer. Despite the low incidence of trisomy 9 mosaicity, patients with this condition are still at risk of aspiration due to gastroesophageal reflux. Patients who survive longer will likely require further management and education regarding their condition.15
We report a case of a male infant diagnosed with trisomy 9 mosaicism who presented with IUGR, facial dysmorphism, and cardiac malformation. Initially, routine peripheral blood karyotype tests did not show any abnormalities; however, the patient was eventually diagnosed with an abnormal finding after performing a cytogenetic microarray test.
Go to :
 XML Download
XML Download